Virus evolution and the predictability of next year's flu
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201705_Bethe.html
Viruses
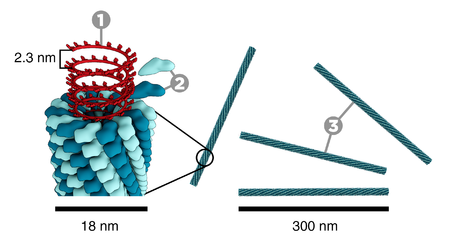 tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
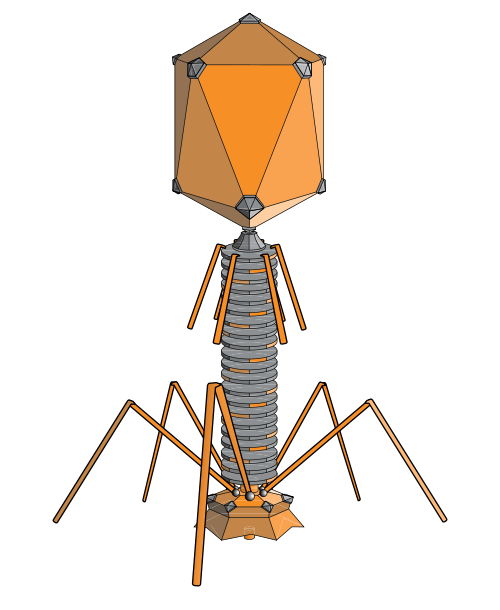
bacteria phage (adenosine, wikipedia)
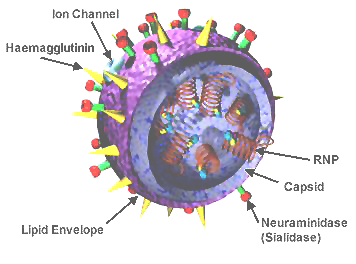
influenza virus wikipedia

human immunodeficiency virus wikipedia
- rely on host to replicate
- little more than genome + capsid
- genomes typically 5-200k bases (+exceptions)
- many infectious diseases are caused by viruses
- very important function in microbial eco-systems
- most abundant organisms on earth $\sim 10^{31}$
Evolution of HIV
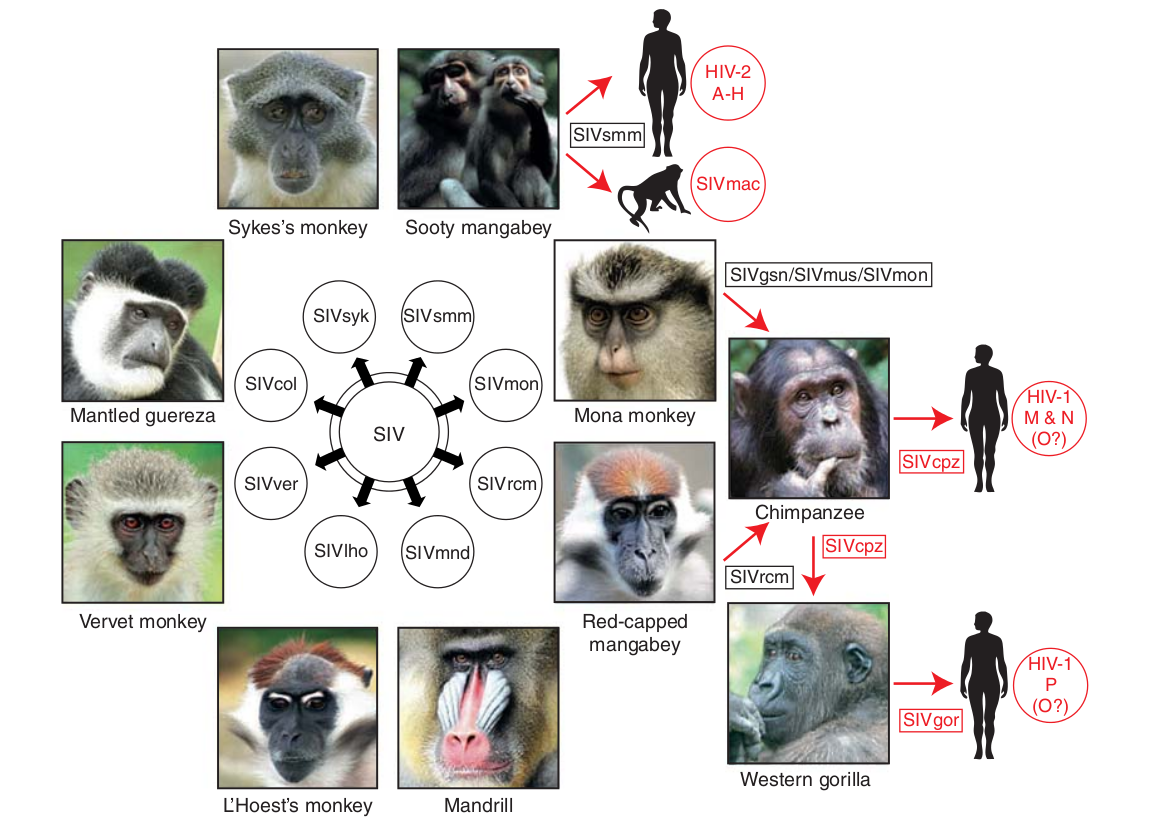
- Chimp → human transmission around 1900 gave rise to HIV-1 group M
- ~100 million infected people since
- subtypes differ at 10-20% of their genome
- HIV-1 evolves ~0.1% per year
HIV infection
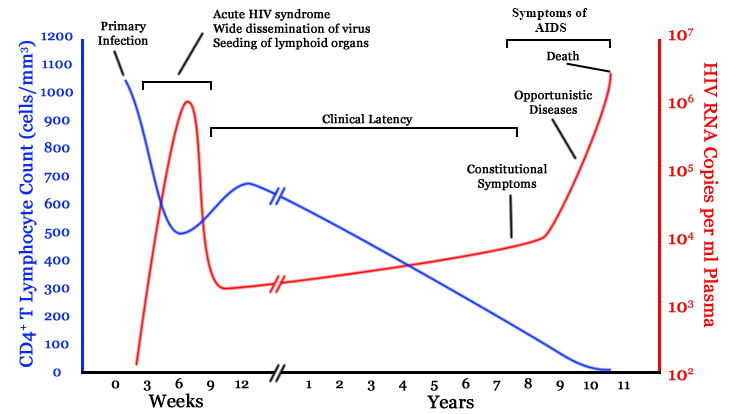

- $10^8$ cells are infected every day
- the virus repeatedly escapes immune recognition
- integrates into T-cells as
latent provirus
{kind=link}
HIV-1 evolution within one individual

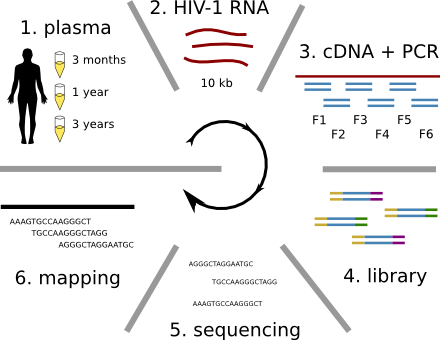

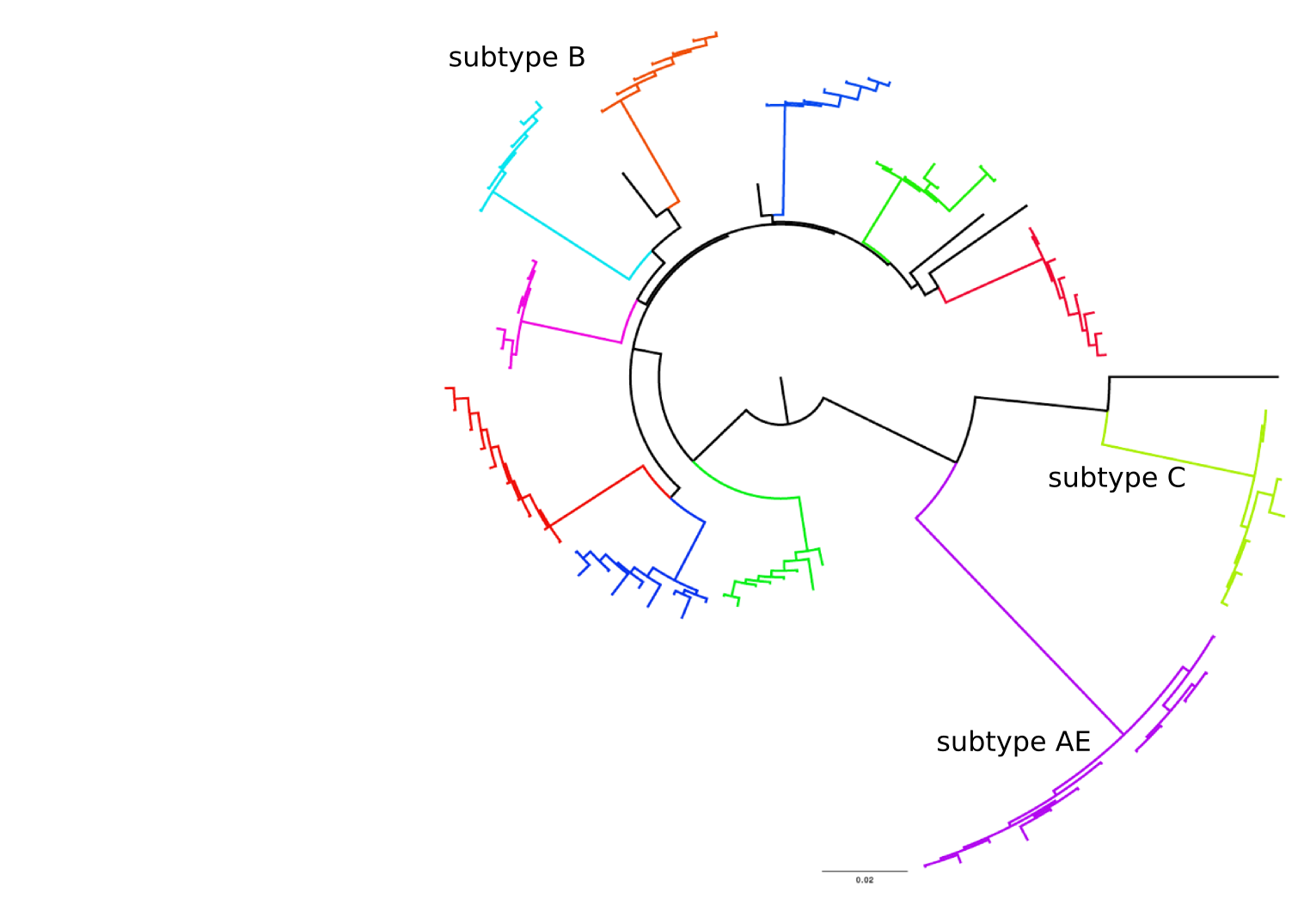
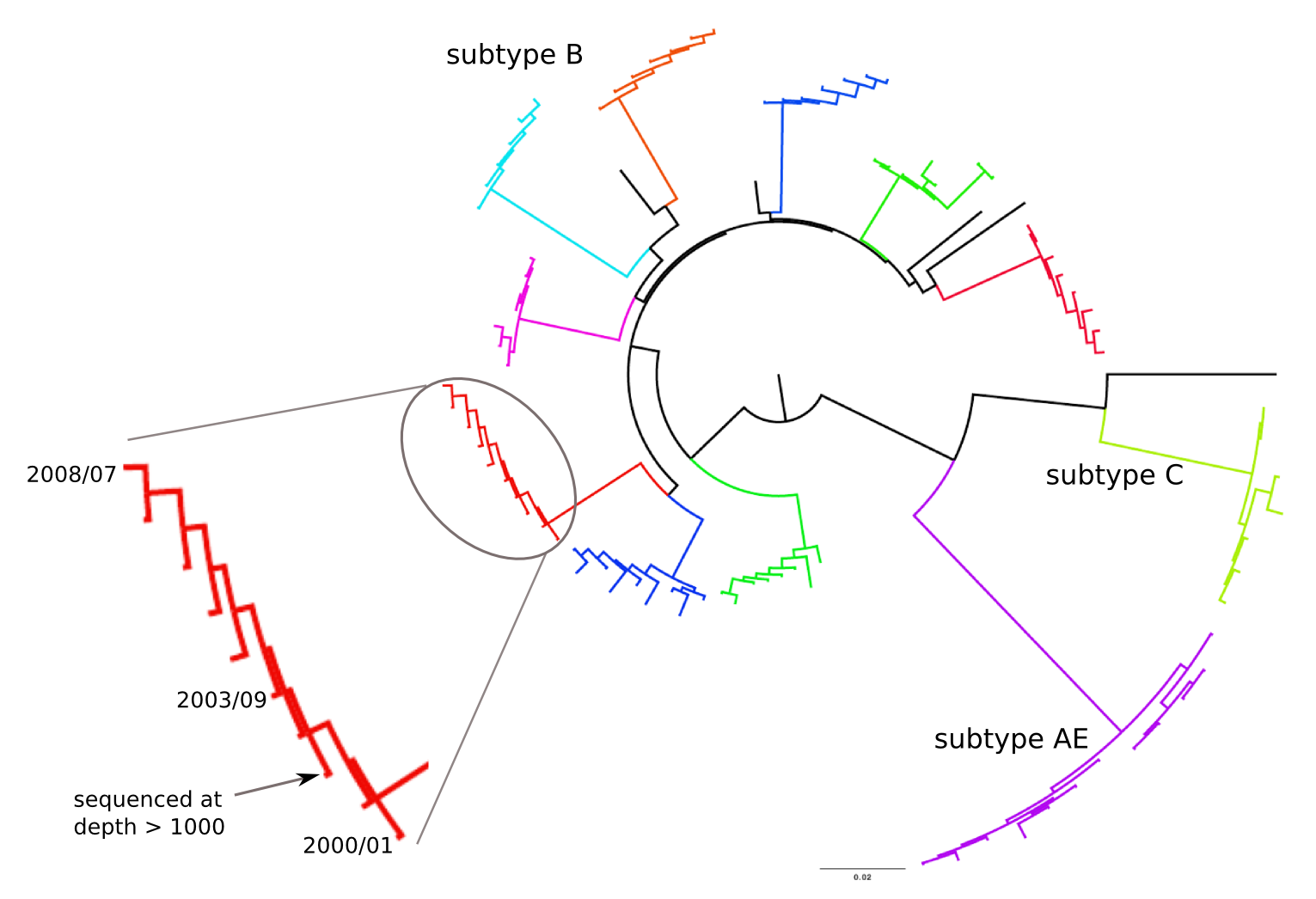
Clonal interference and traveling waves
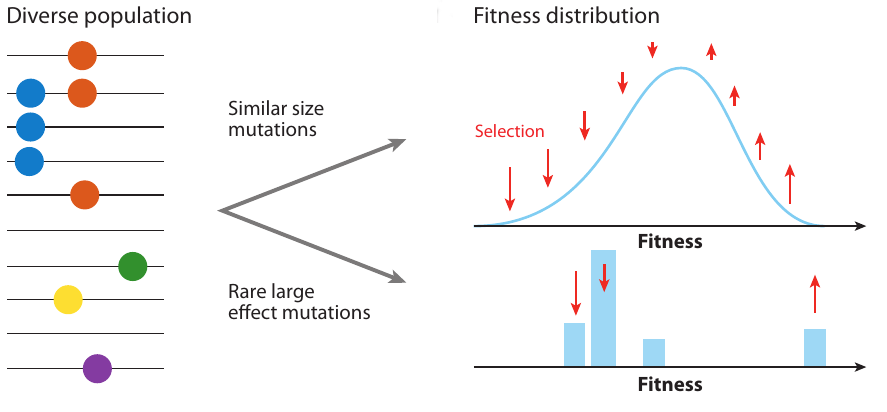
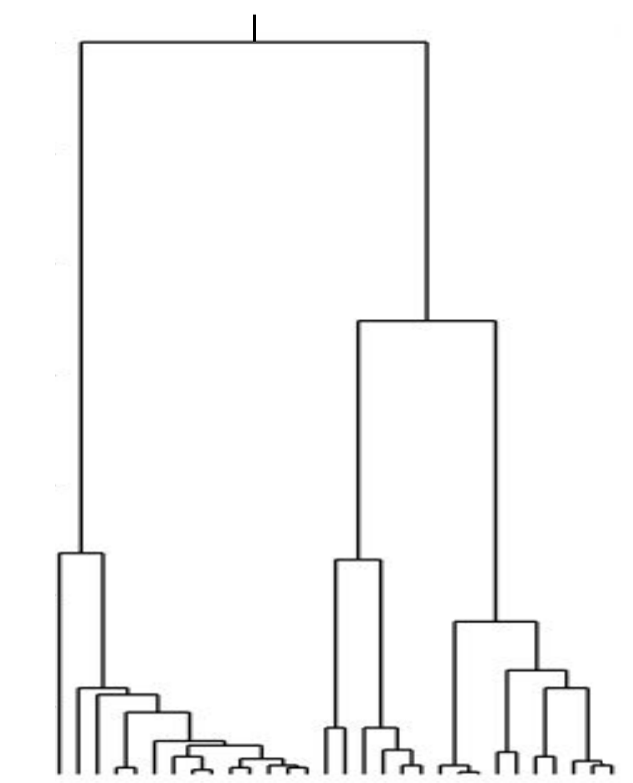
Neutral/Kingman coalescent
strong selection
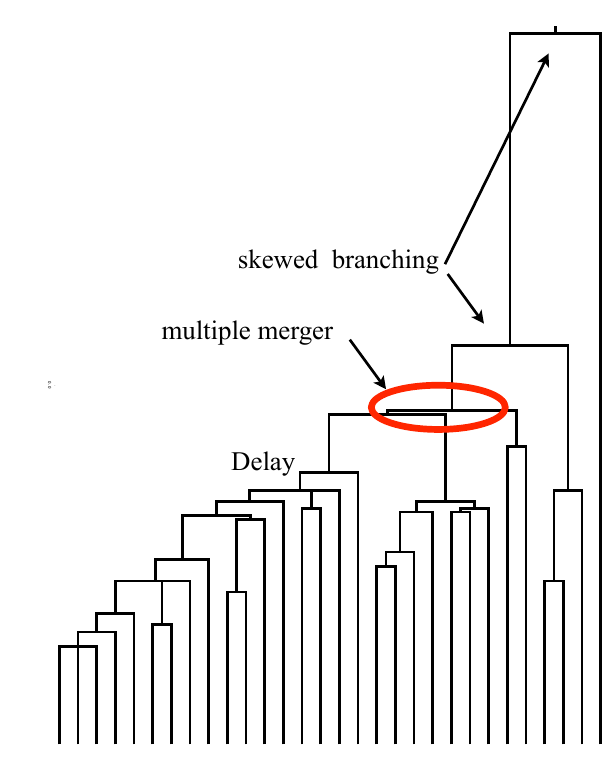
Bolthausen-Sznitman Coalescent
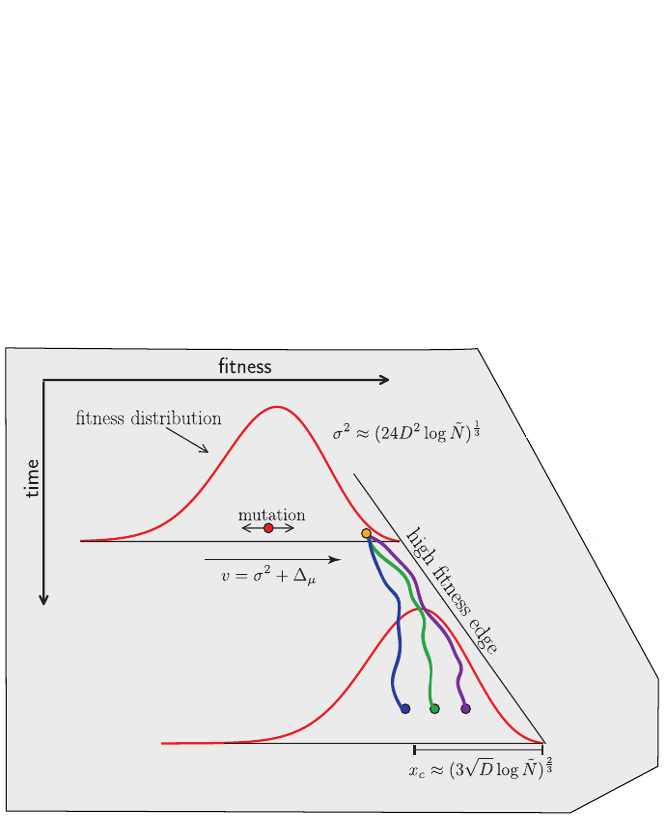
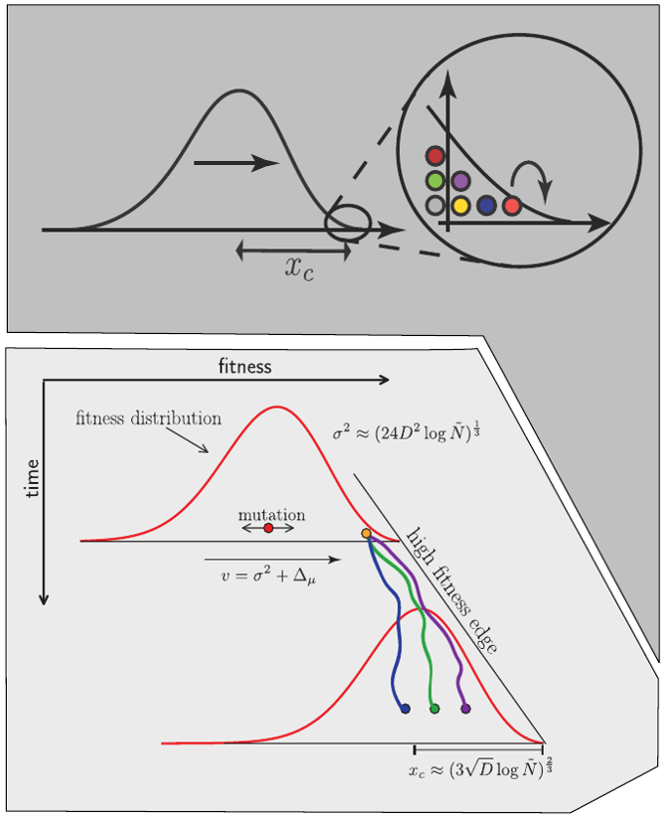
Traveling waves and the Bolthausen-Snitman coalescent
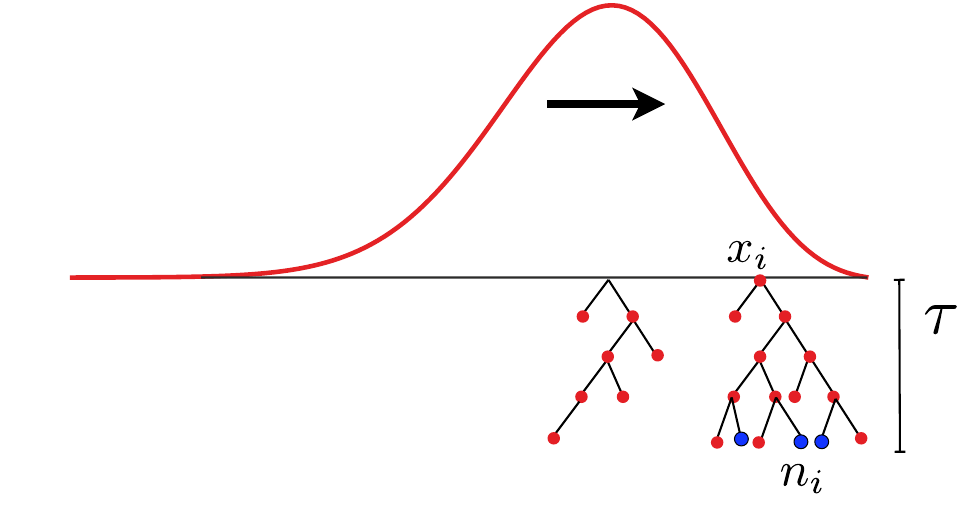
- Branching process approximation: $P(n_i, t|x_i)$
-
Does a sample (blue dots) have a common ancestor $\tau$ generations ago?
$\quad Q_b = \langle \sum_i \left(\frac{n_i}{\sum_j n_j}\right)^b\rangle \approx \frac{\tau-T_c}{T_c(b-1)} $ -
All other merger rates are also consistent with the Bolthausen-Sznitman coalescent:
$\quad\lambda_{b,k} = \frac{(k-2)!(b-k)!}{T_c (b-1)!}$
U-shaped polarized site frequency spectra
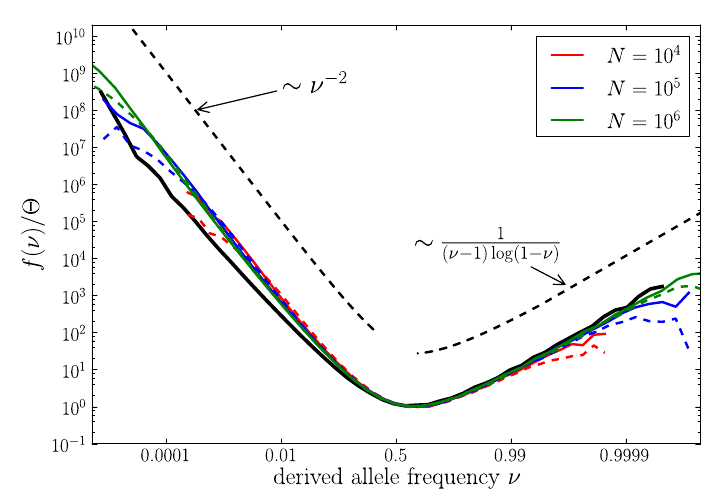

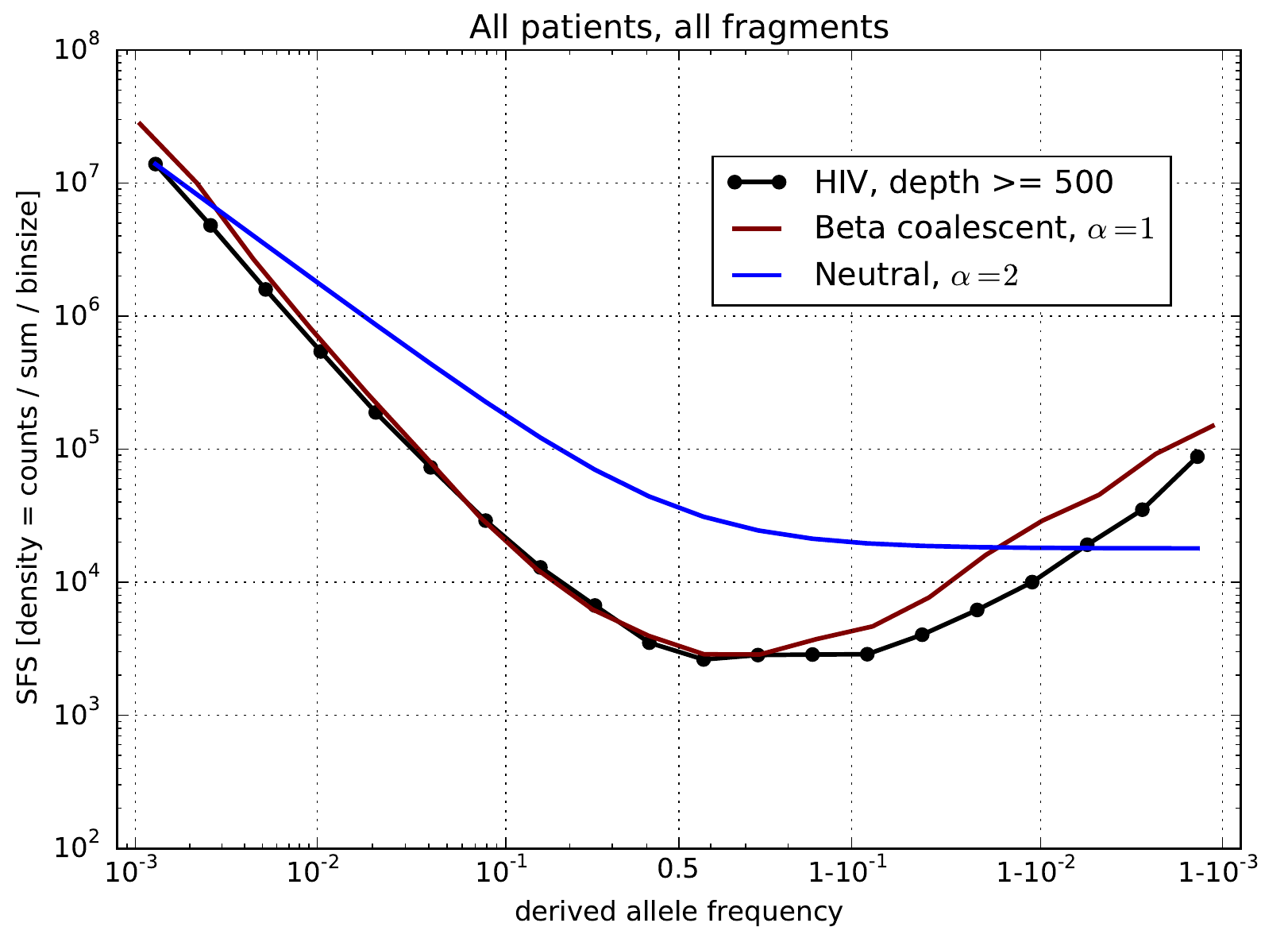 Zanini et al, eLife, 2015
Zanini et al, eLife, 2015
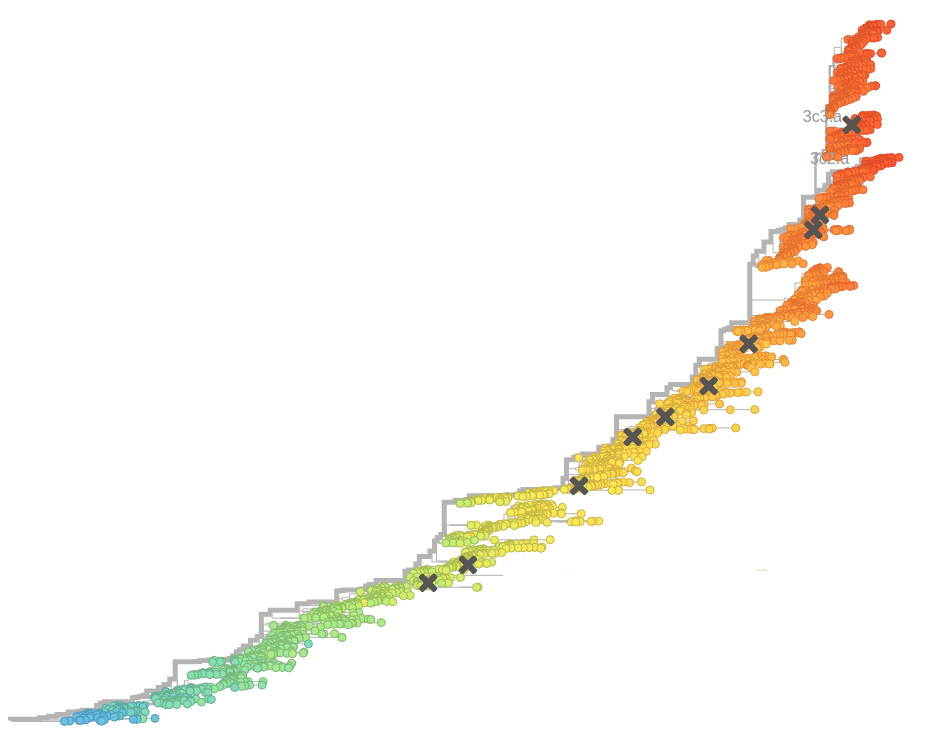
Bursts in a tree ↔ high fitness genotypes
Can we read fitness of a tree?
Human seasonal influenza viruses
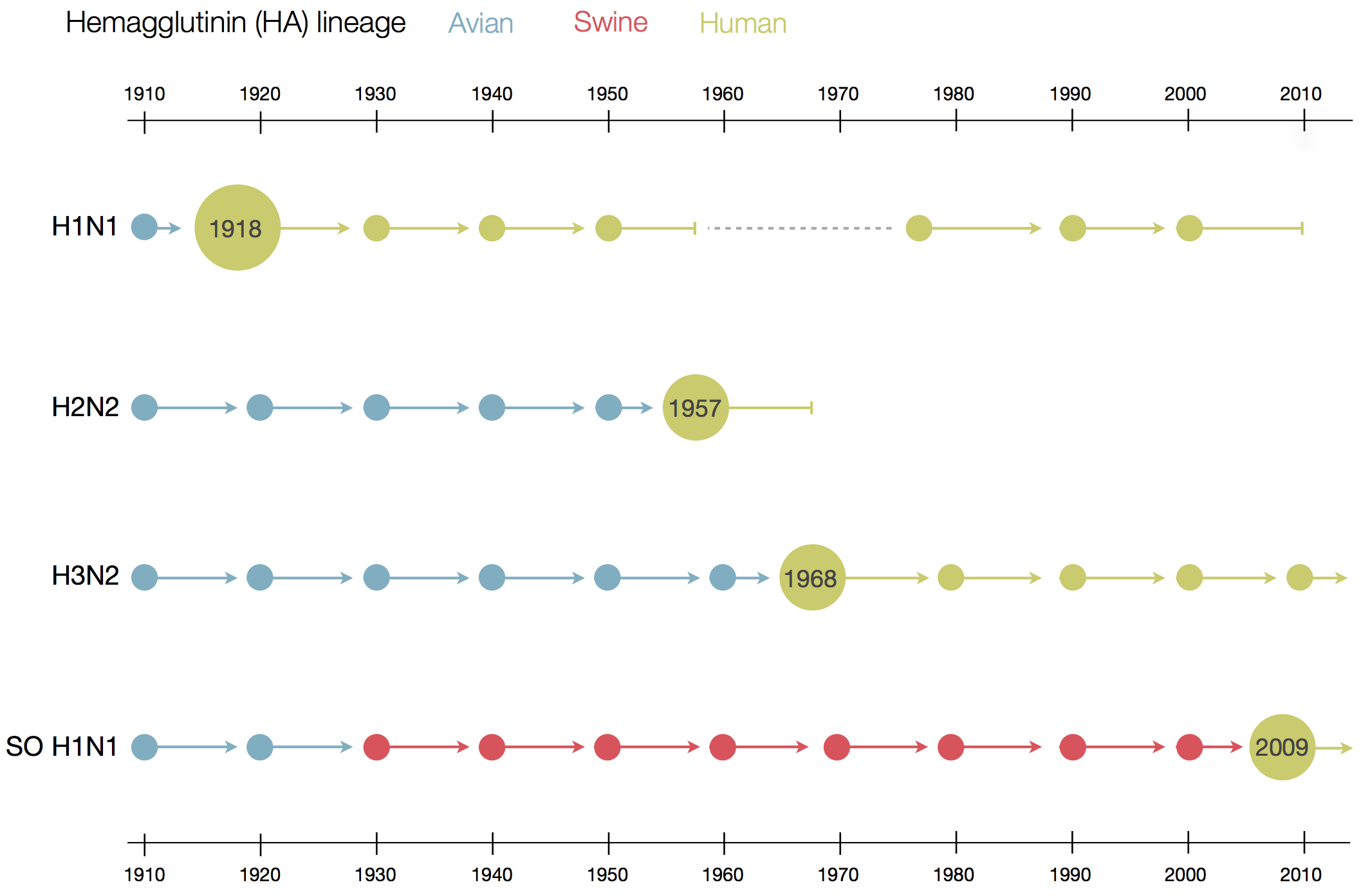
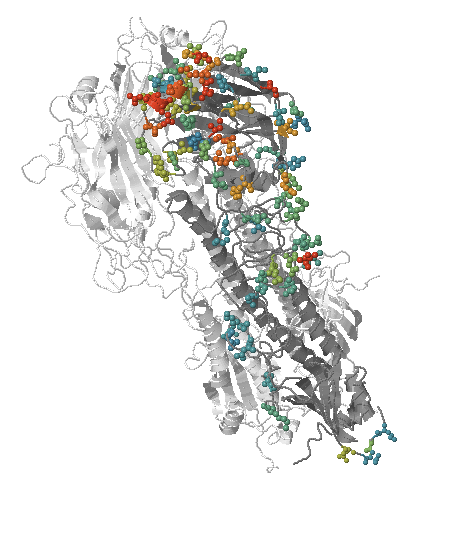
- Influenza virus evolves to avoid human immunity
- Vaccines need frequent updates
Predicting evolution
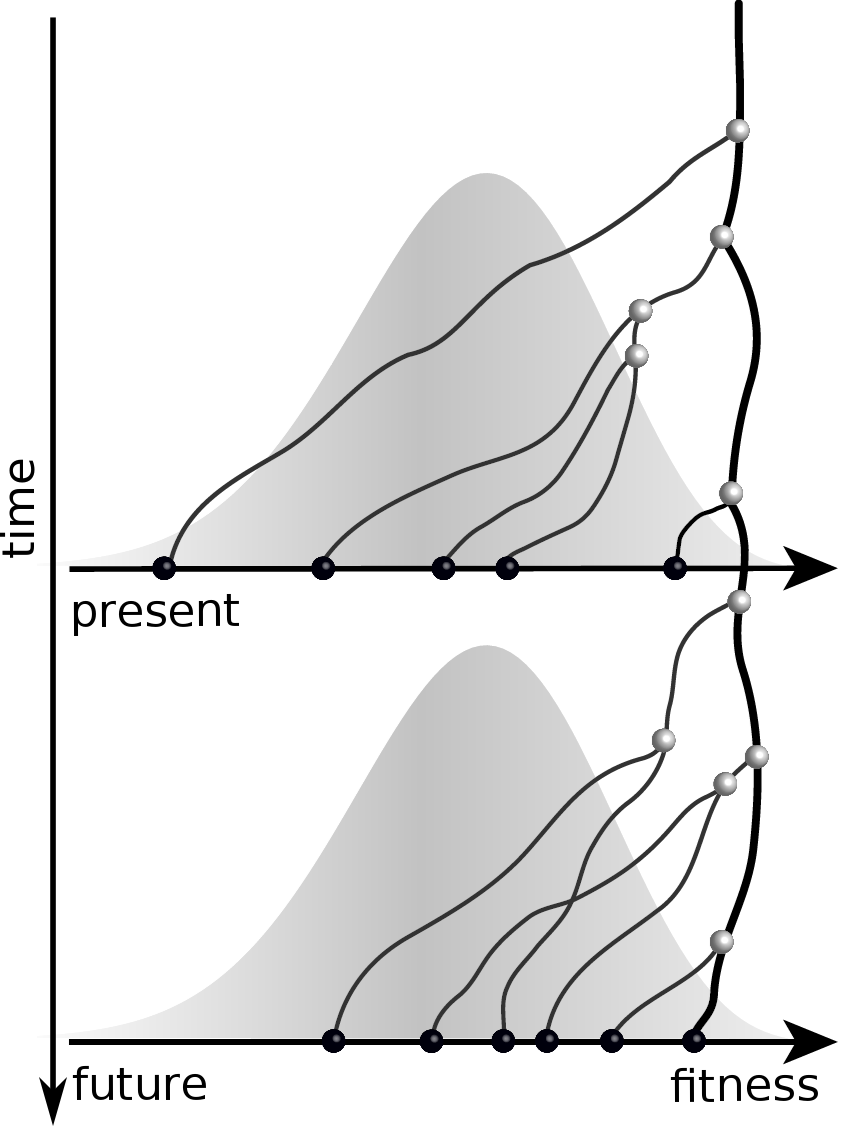
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
Validate on simulation data
- simulate evolution
- sample sequences
- reconstruct trees
- infer fitness
- predict ancestor of future
- compare to truth
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
nextstrain.org
Summary
- RNA virus evolution can be observed directly
- Extensive reversion to preferred amino acid sequence
- Rapidly adapting population require new population genetic models
- Those model can be used to infer fit clades
- Future influenza population can be anticipated
- Automated real-time analysis can help fight the spread of disease
HIV acknowledgments
- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
nextstrain.org
- Trevor Bedford
- Colin Megill
- Pavel Sagulenko
- Wei Ding
- Sidney Bell
- James Hadfield
