Real-time analysis to track and predict pathogen spread
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201709_USB_NGS.html
Outbreaks require a rapid and informed response
- influenza virus (spanish flu 1918, "swine flu" 2009, H5N1, ...)
- SARS (Severe Acute Respiratory Syndrome, coronavirus)
- MERS (Middle East respiratory syndrome, coronavirus)
- Ebola (filovirus)
- Zika virus (flavivirus)
- ...
Human seasonal influenza viruses
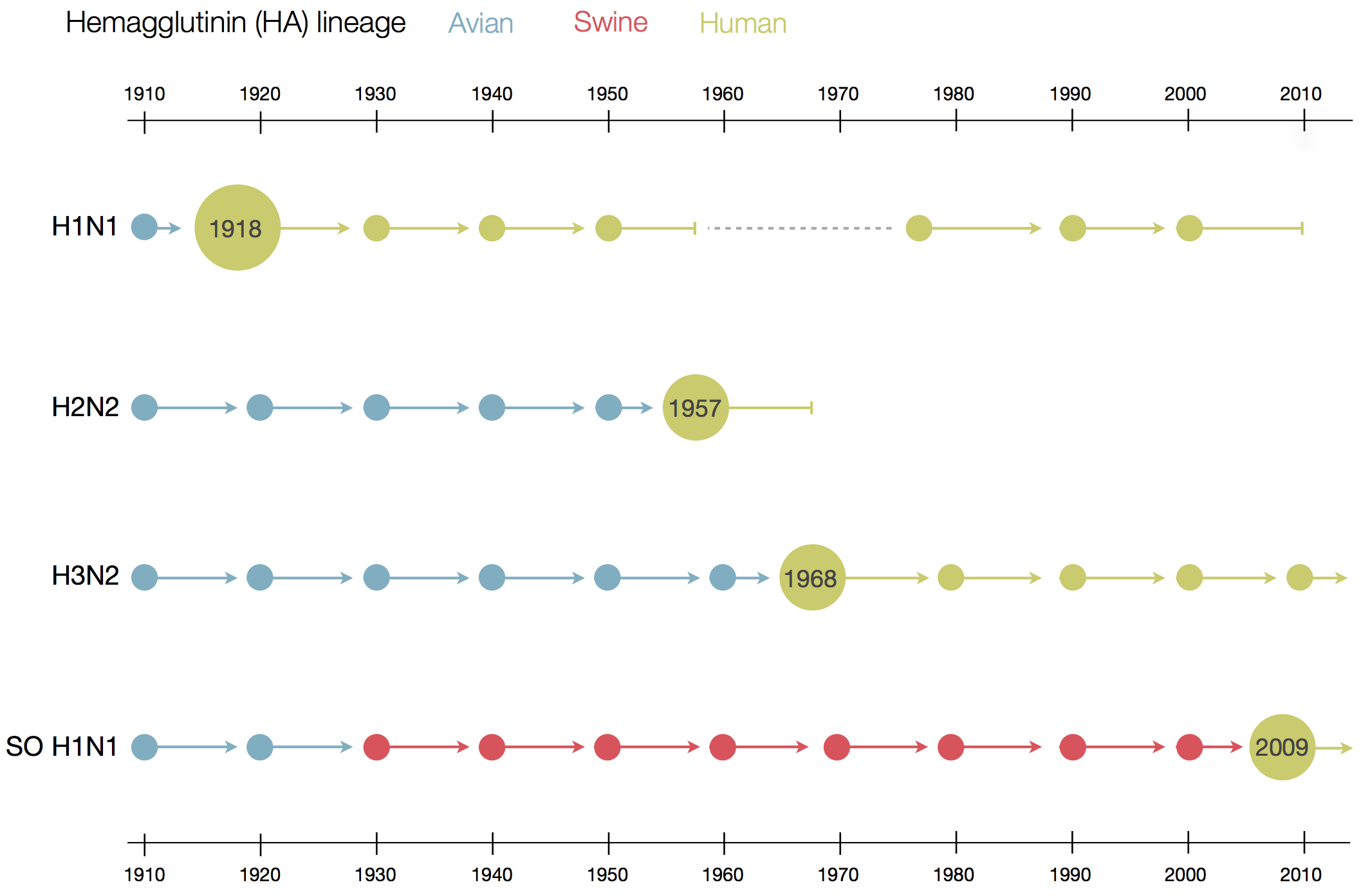
Surveillance of human seasonal influenza viruses
- WHO CCs and NICs sequence and phenotype 100s of viruses per month
- Sequences allow us to track how the virus is spreading and changing
nextflu.org
joint work with Trevor Bedford & his lab
Ad-hoc response to other diseases
- Outbreaks occur in unpredictable places
- Can spread across the globe in weeks
- Different pathogens require different microbiological expertise
- Sequencing is a universal way to investigate early spread and source of the pathogen
Challenges in rapid outbreak sequencing
- sample → DNA to sequence
- sequencing itself not too big a problem
- combining data from different sources
- convincing groups to share and pool their data
- rapid analysis and dissemination of results
 Raymond Koundouno, image by Sophie Darrafour
Raymond Koundouno, image by Sophie Darrafour
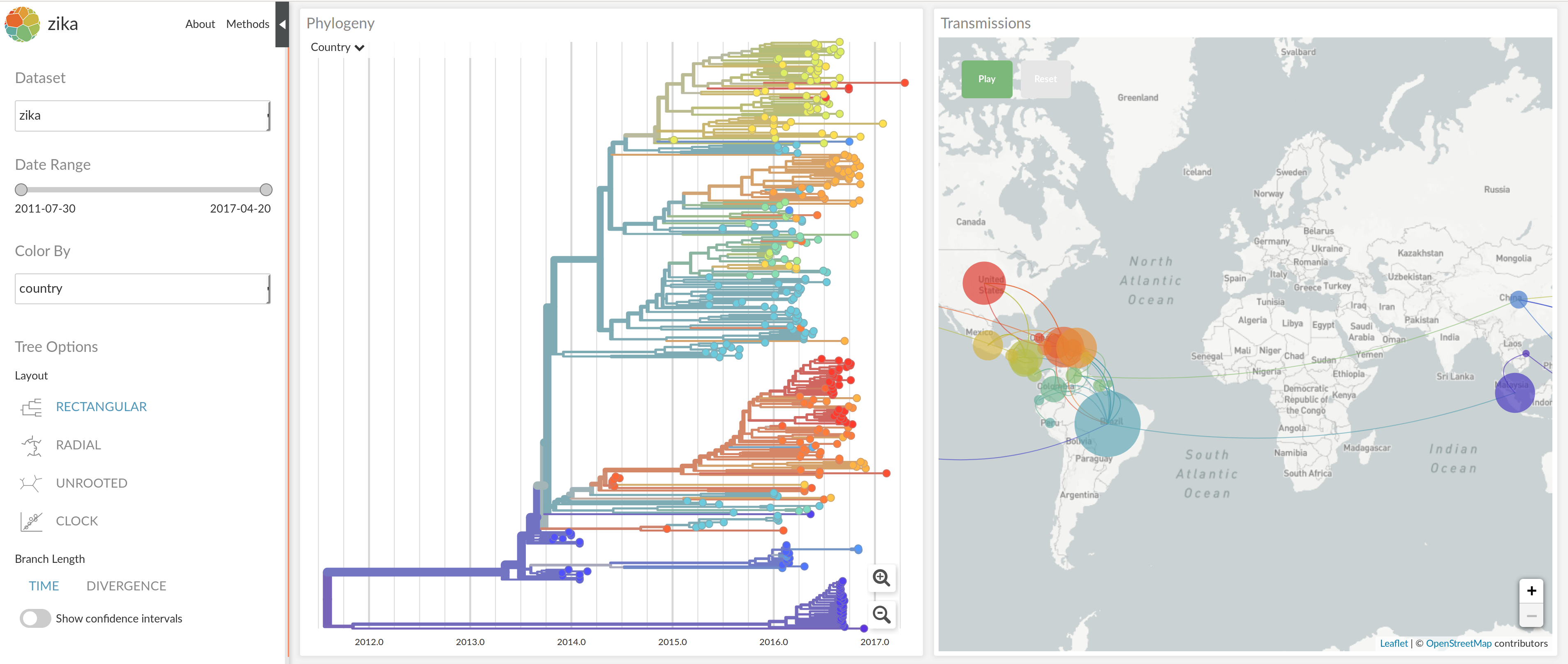
nextstrain.org
joint work with Trevor Bedford & his lab
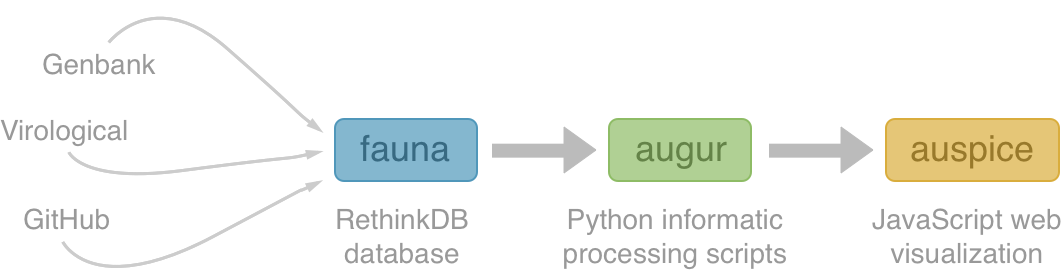
NextStrain architecture
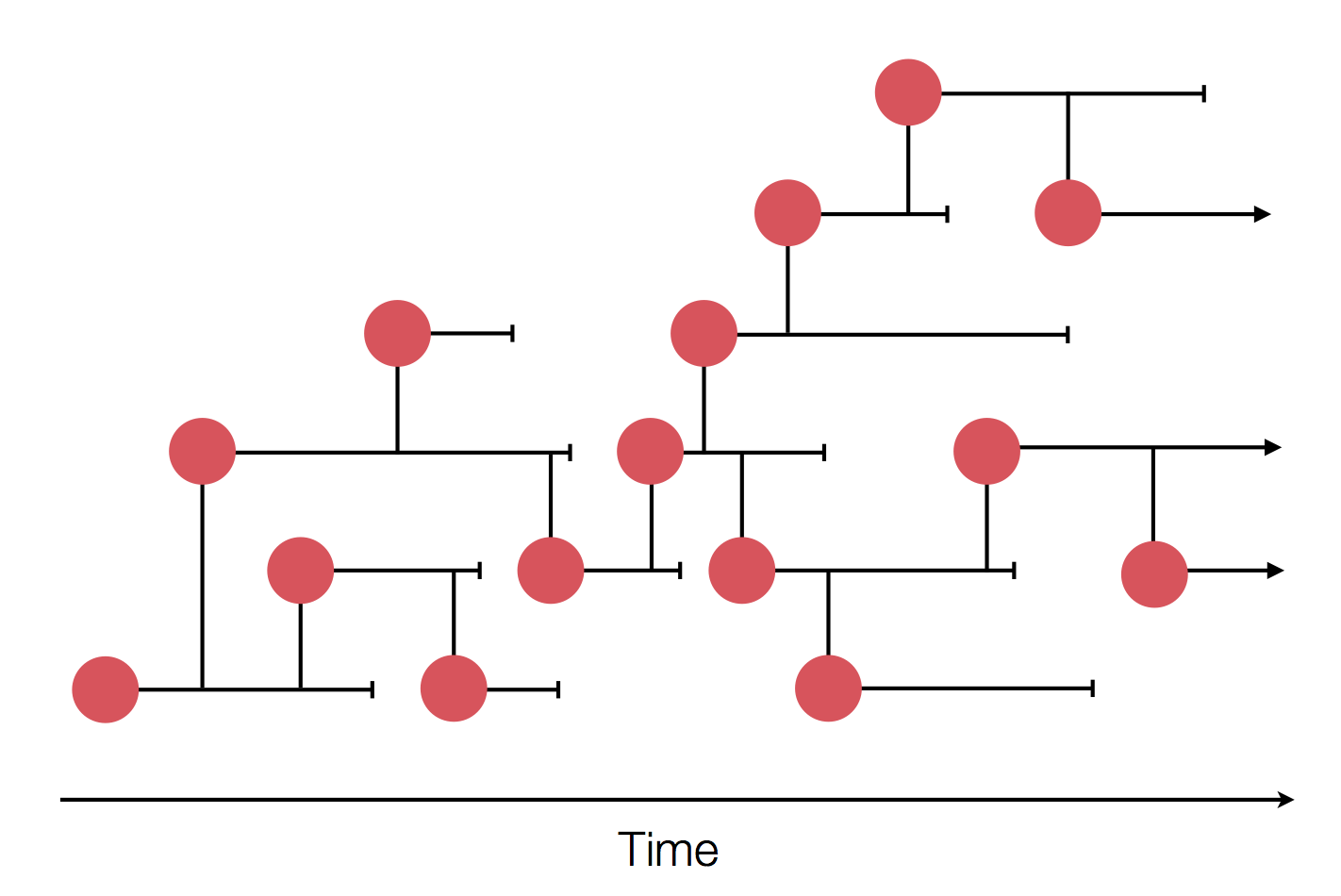
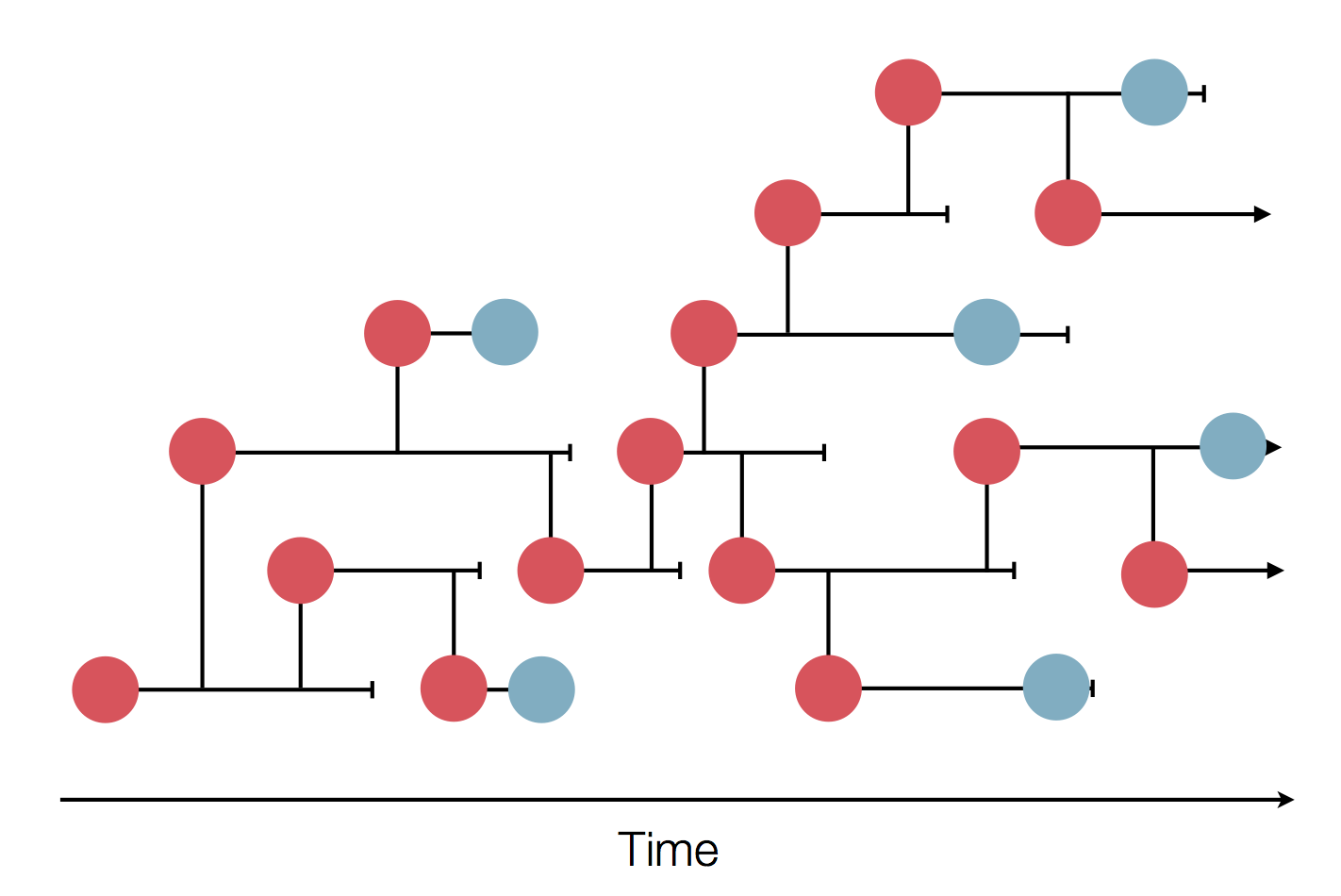
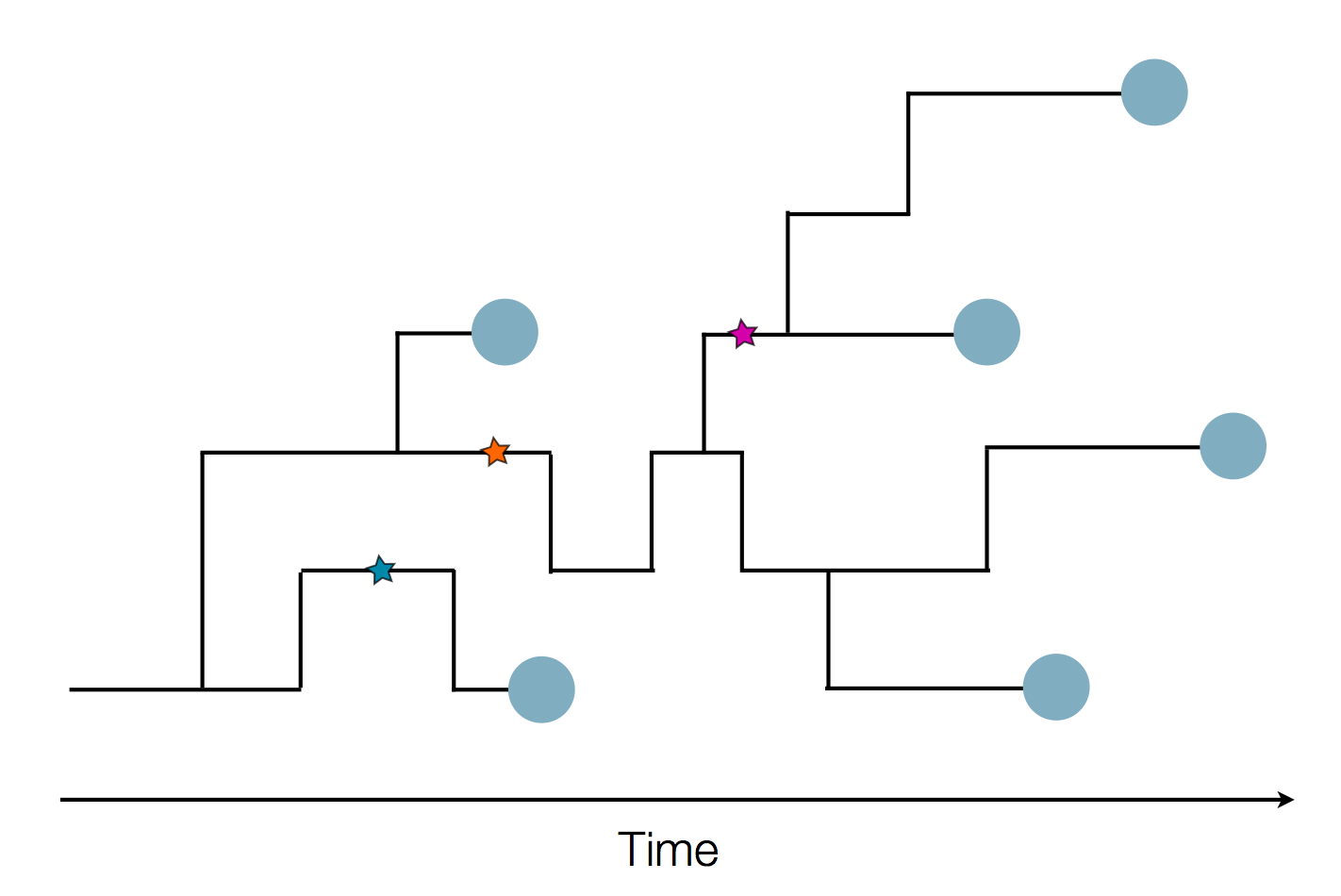
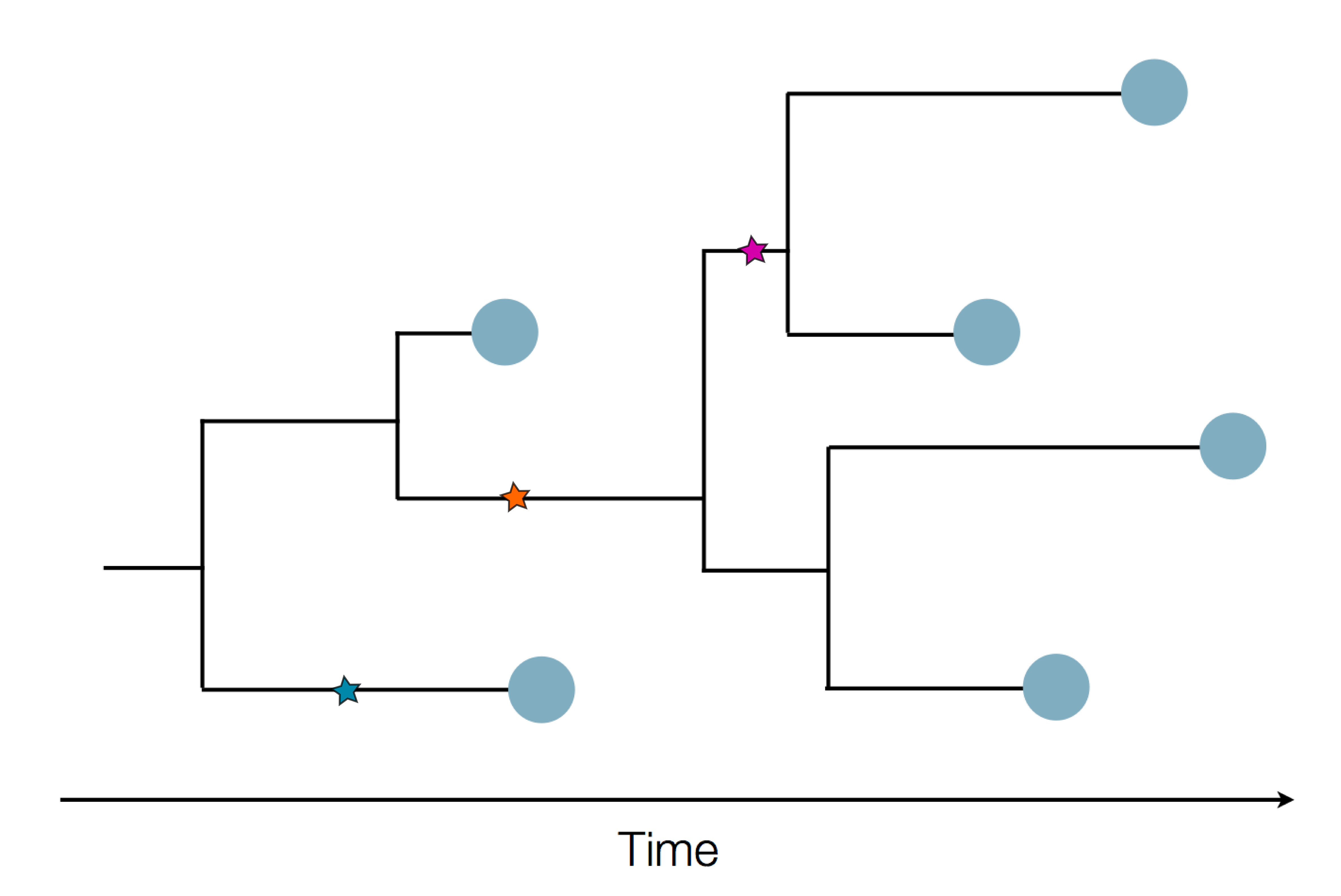
Using treetime to rapidly compute timetrees
Why are bacteria harder?
- Much larger genomes
- Slower evolution
- horizontal transfer
- The most important parts are often hardest to analyze
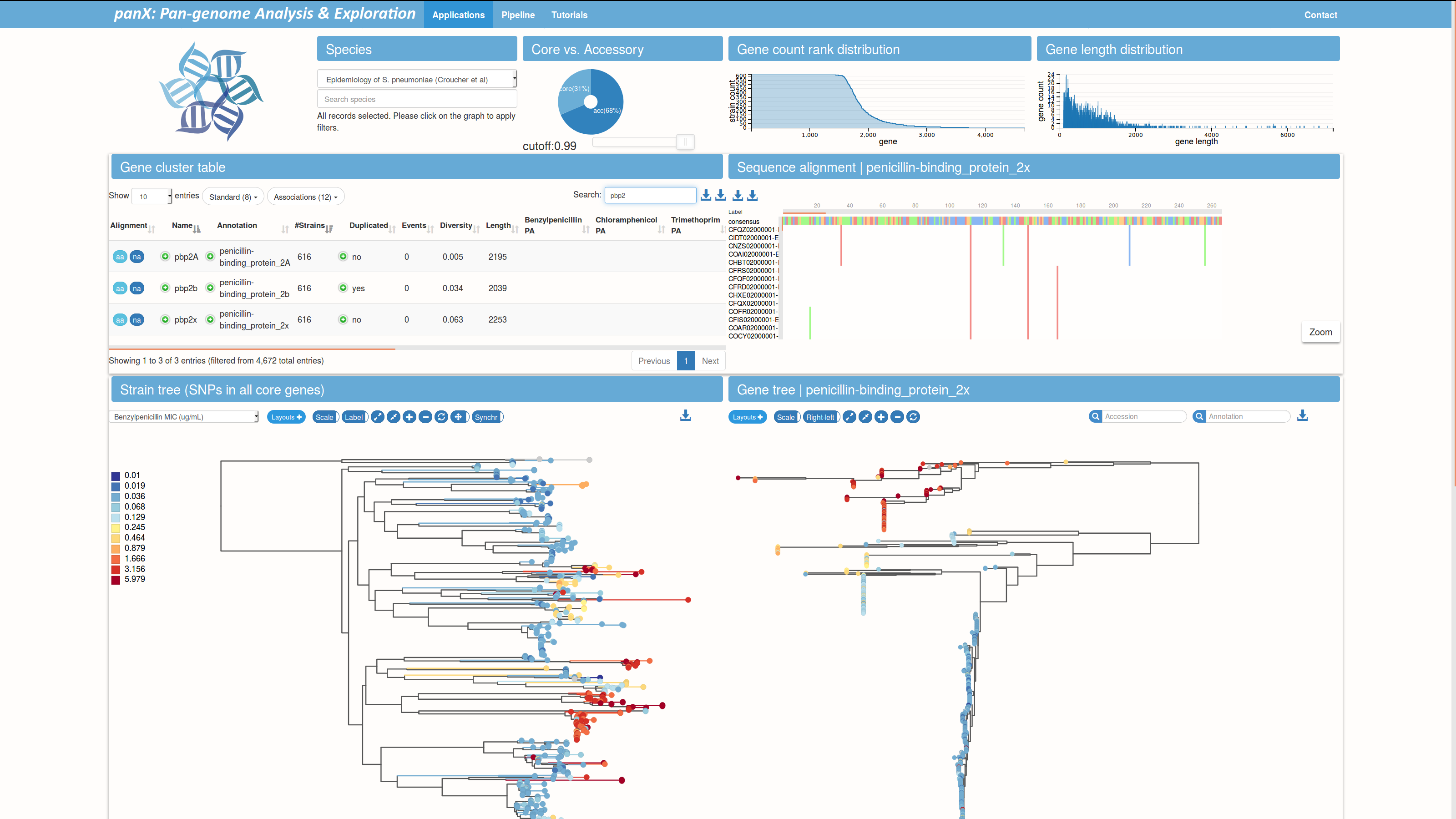
Pan-genome analysis of fragmented WGS assemblies
pangenome.de
Virus evolution takes place within the host
Deep longitudinal sampling is necessary to monitor evolution in detail
Evolution of HIV
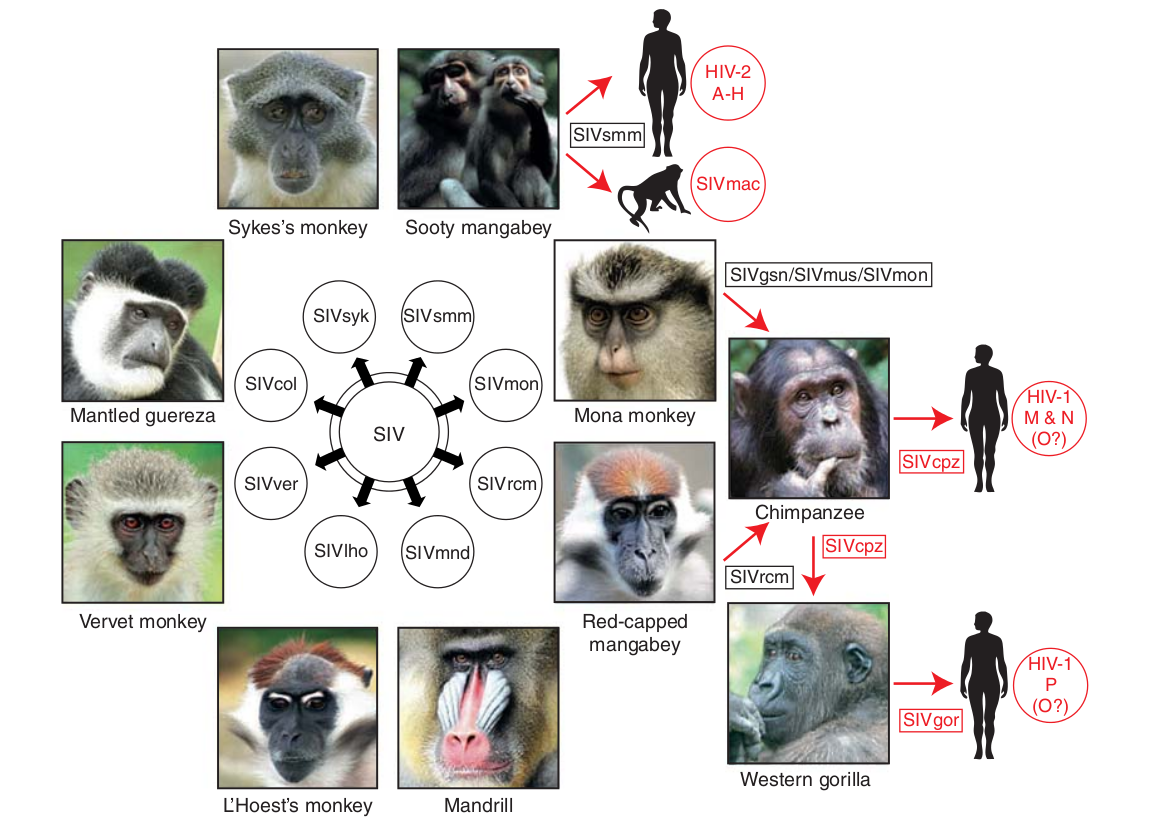
- Chimp → human transmission ~1900 gave rise to HIV-1 group M
- Diversified into subtypes that are ~20% different
- evolves at a rate of about 0.1% per year
HIV-1 evolution within one individual

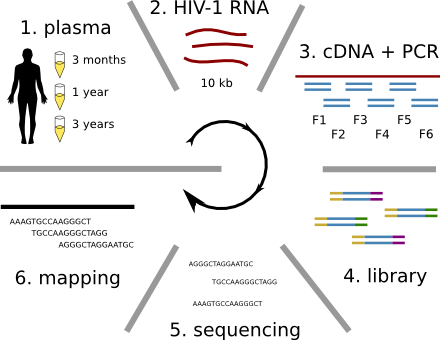

Accuracy of minor variant frequencies

Sharing of HIV-1 data
- NGS data requires extensive filtering/cleaning/processing
- Raw reads in short read archive often not helpful
- Especially true for structured data sets
- hiv.biozentrum.unibas.ch
HIV acknowledgments
- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller
nextstrain.org team and panX
- Colin Megill
- Trevor Bedford
- James Hadfield
- Sidney Bell
- Wei Ding
- Pavel Sagulenko
![]()
![]()
![]()