Virus evolution and the predictability of next year's flu
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201711_NIBR.html
Human seasonal influenza viruses
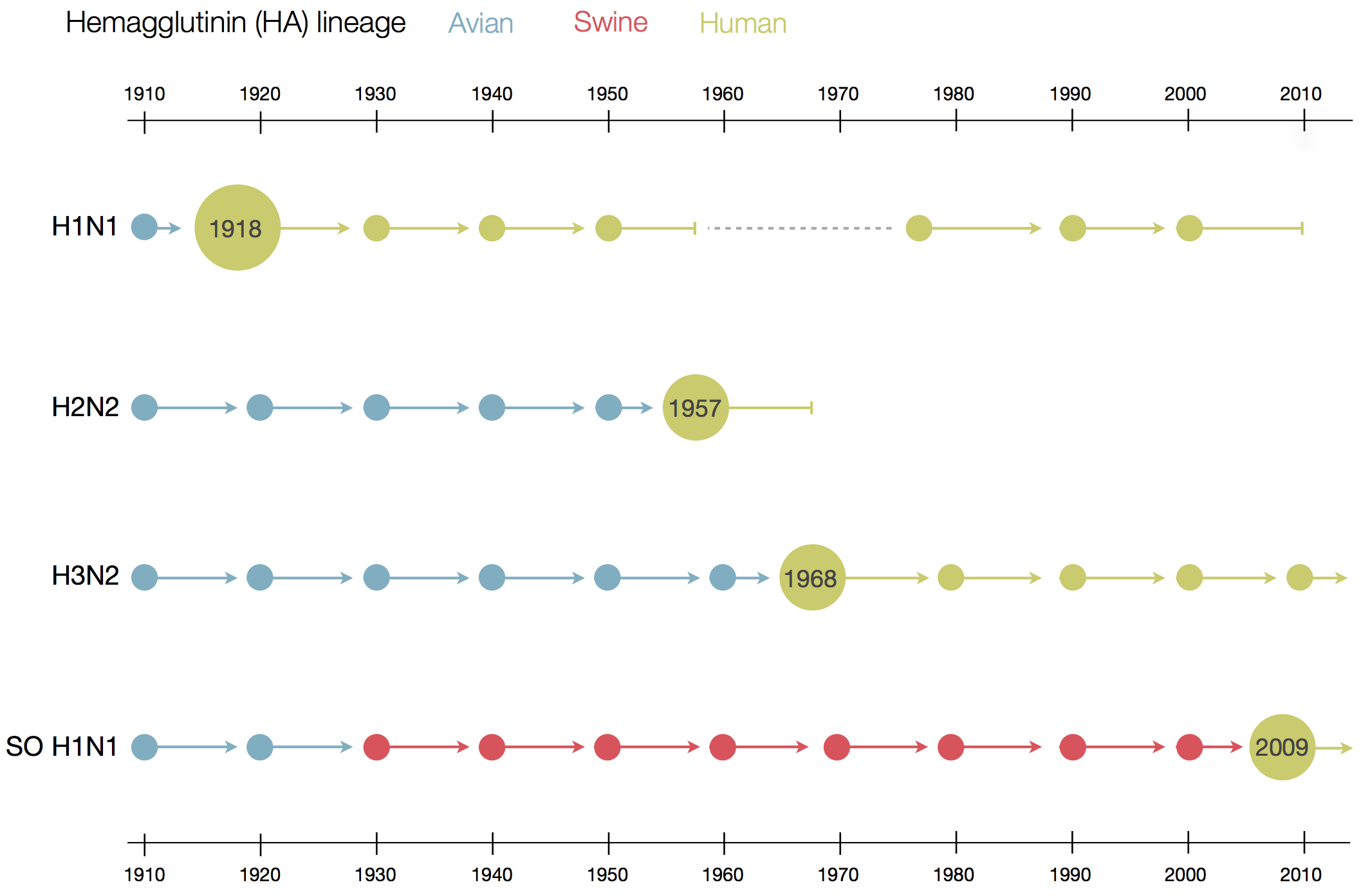
- Influenza virus evolves to avoid human immunity
- Vaccines need frequent updates
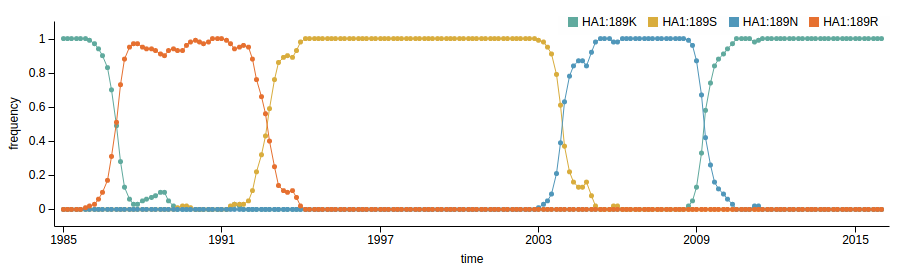
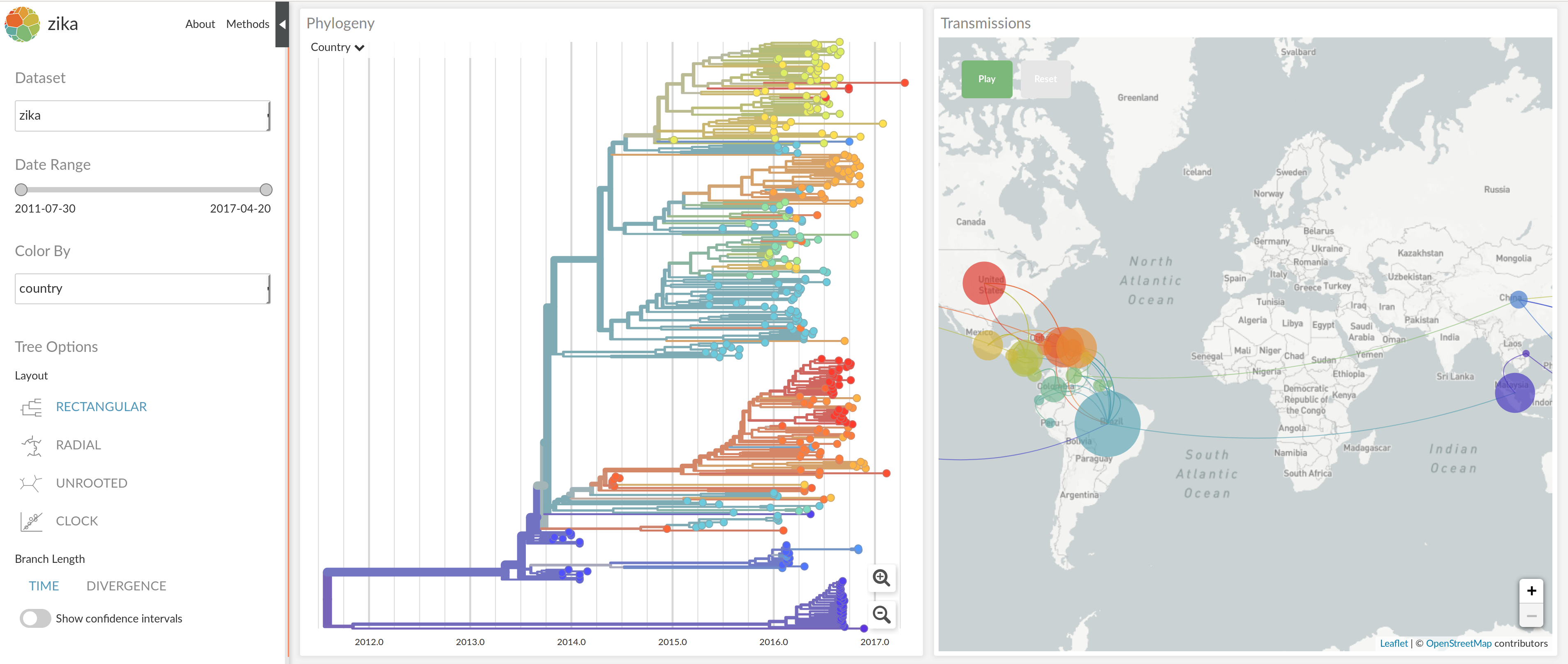
nextflu.org
joint work with Trevor Bedford & his lab
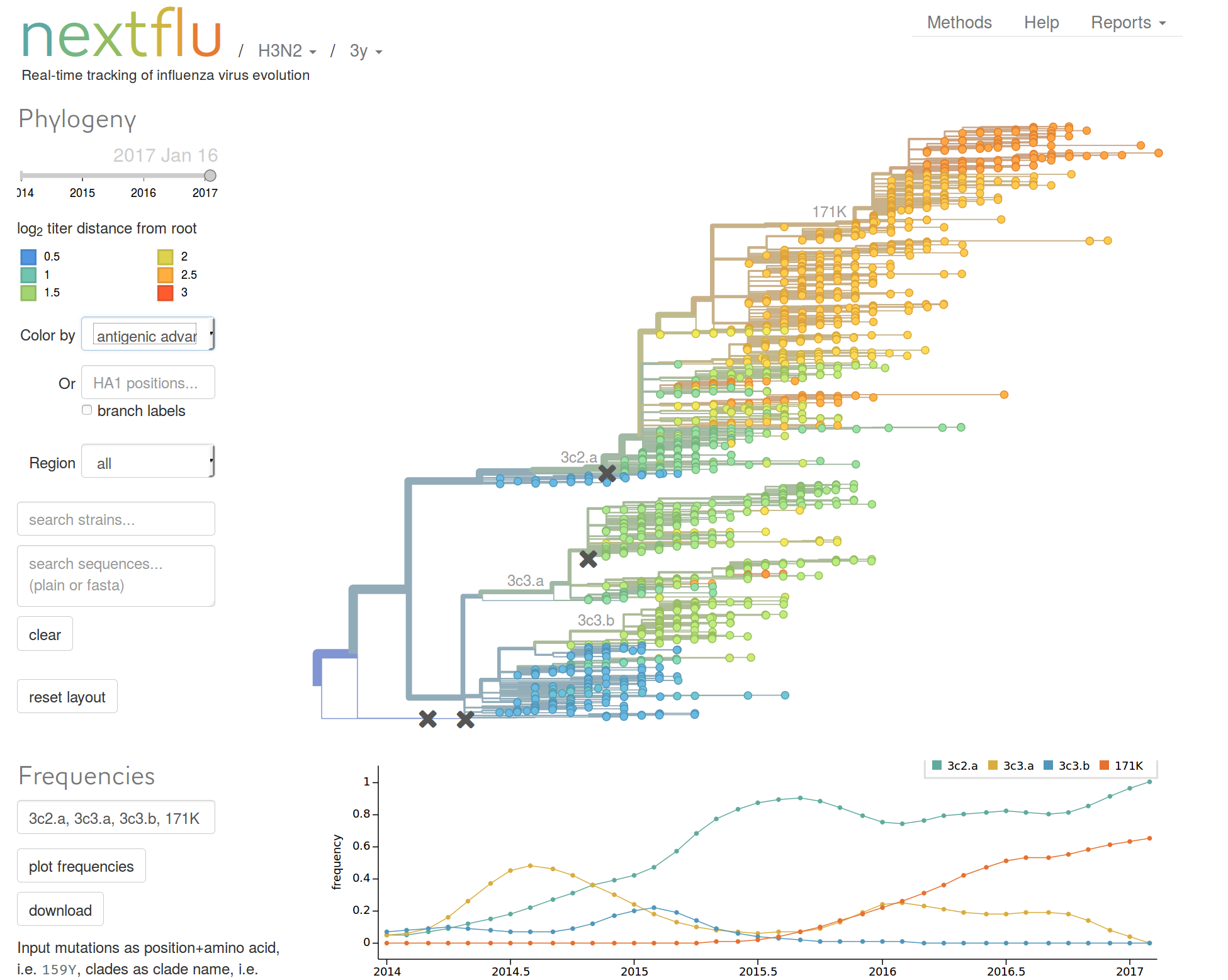
Beyond tracking: can we predict?
Clonal interference and traveling waves
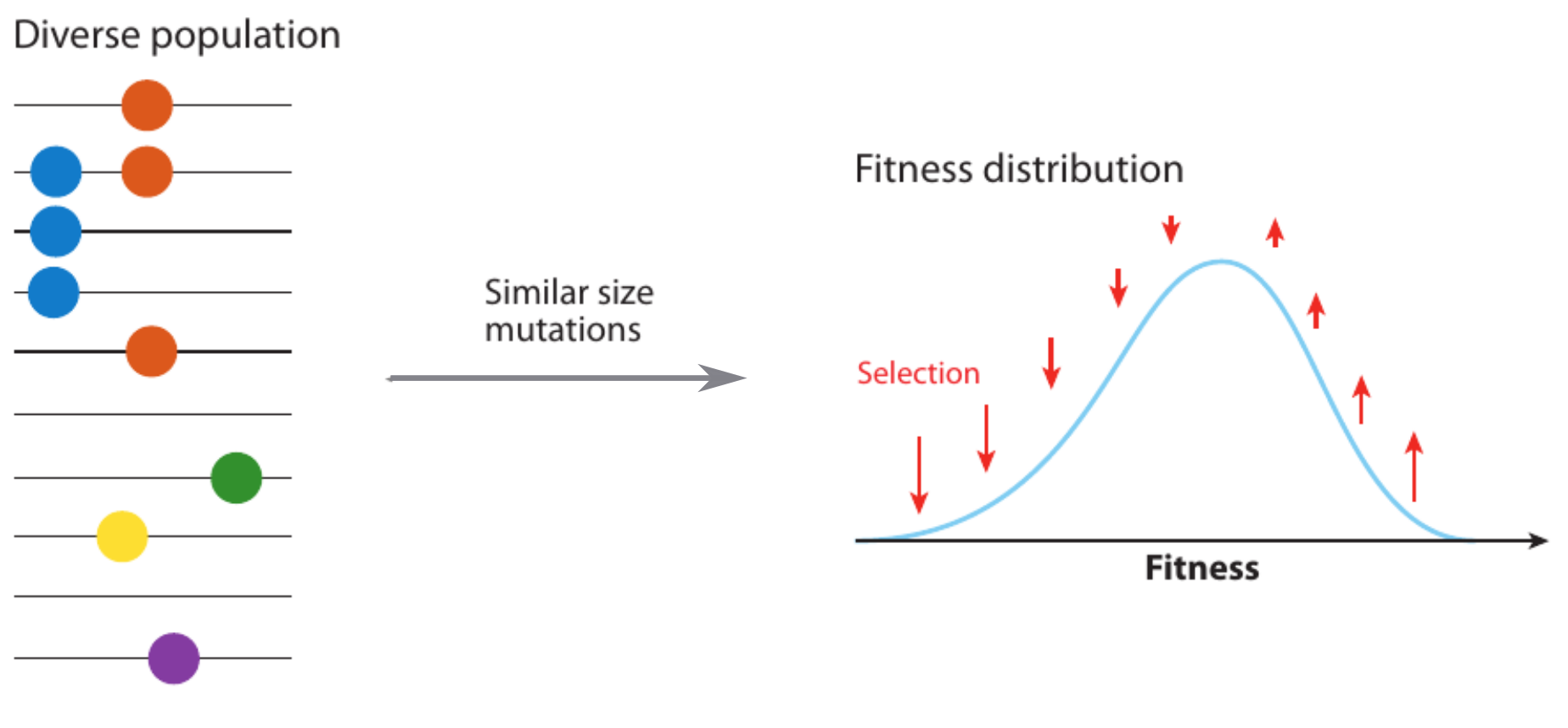
Theoretical framework for virus evolution -- population genetics




evolutionary processes ↔ trees ↔ genetic diversity
Typical tree
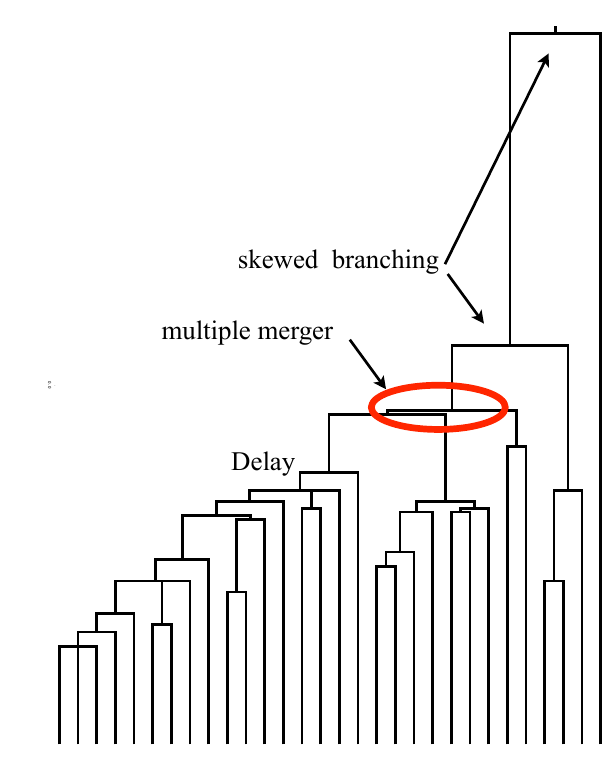
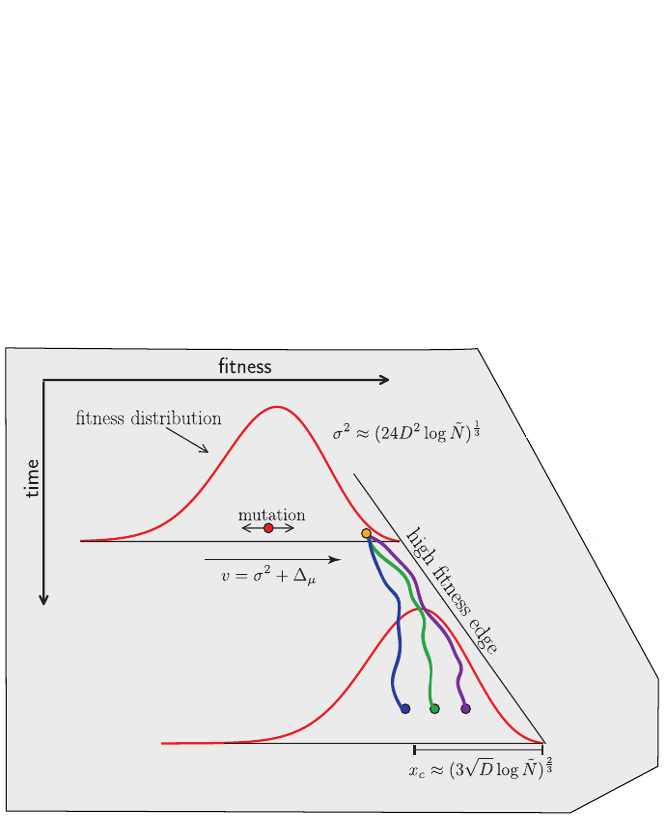
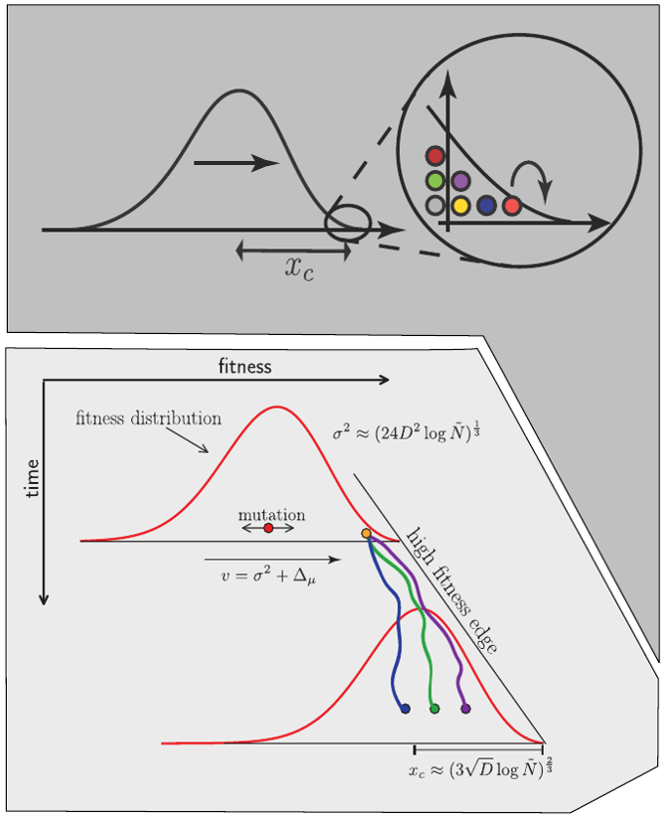
Bolthausen-Sznitman Coalescent
Bursts in a tree ↔ high fitness genotypes
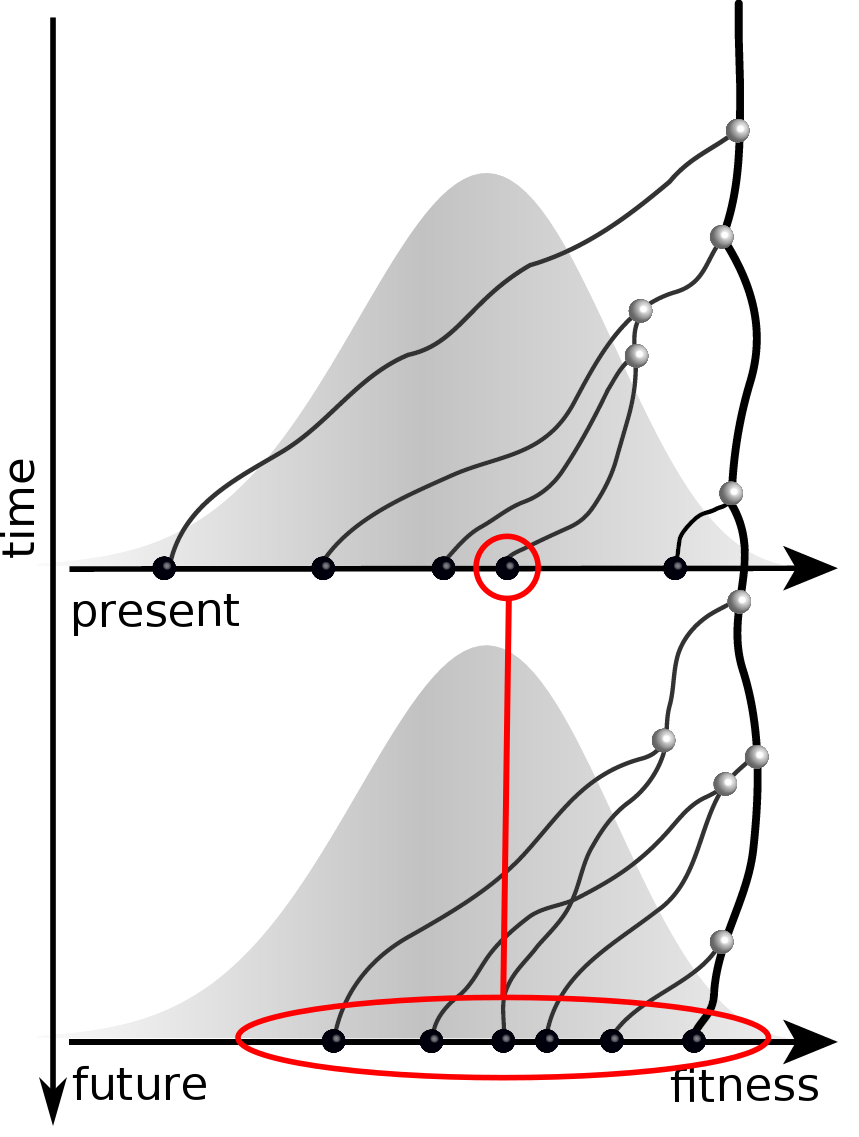
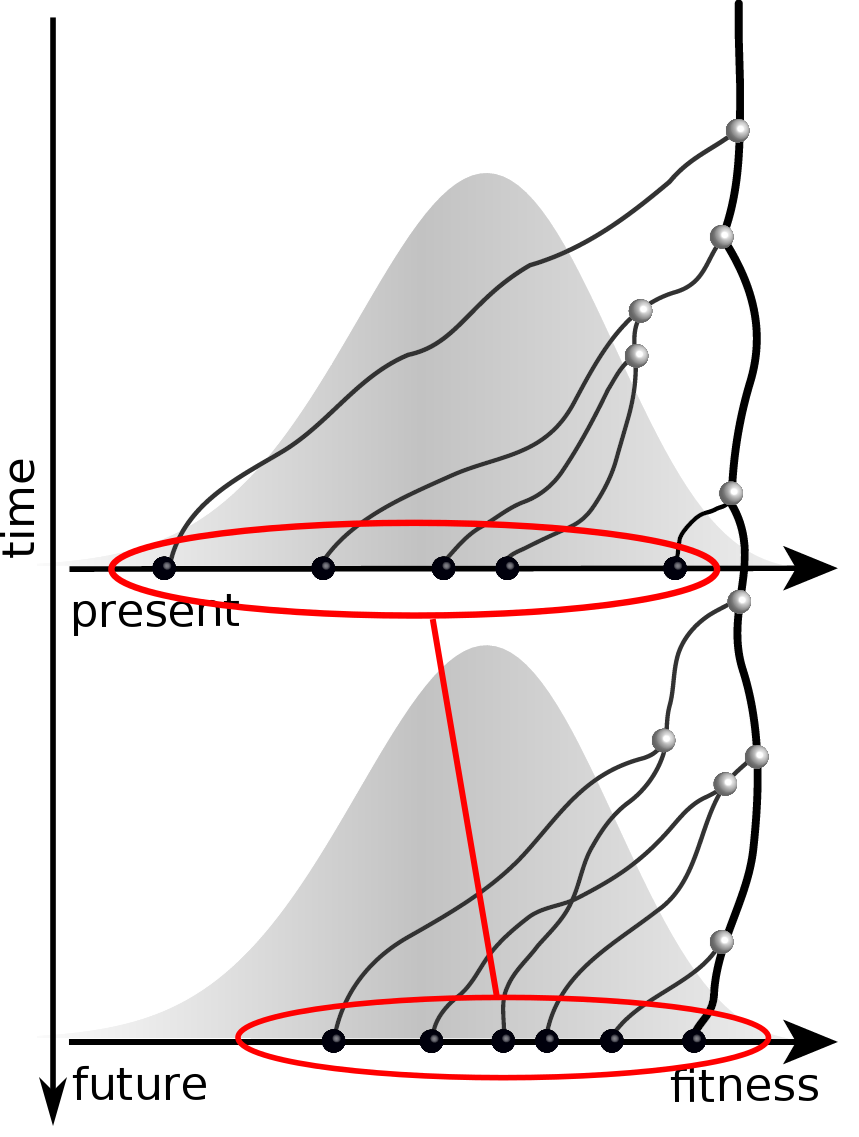
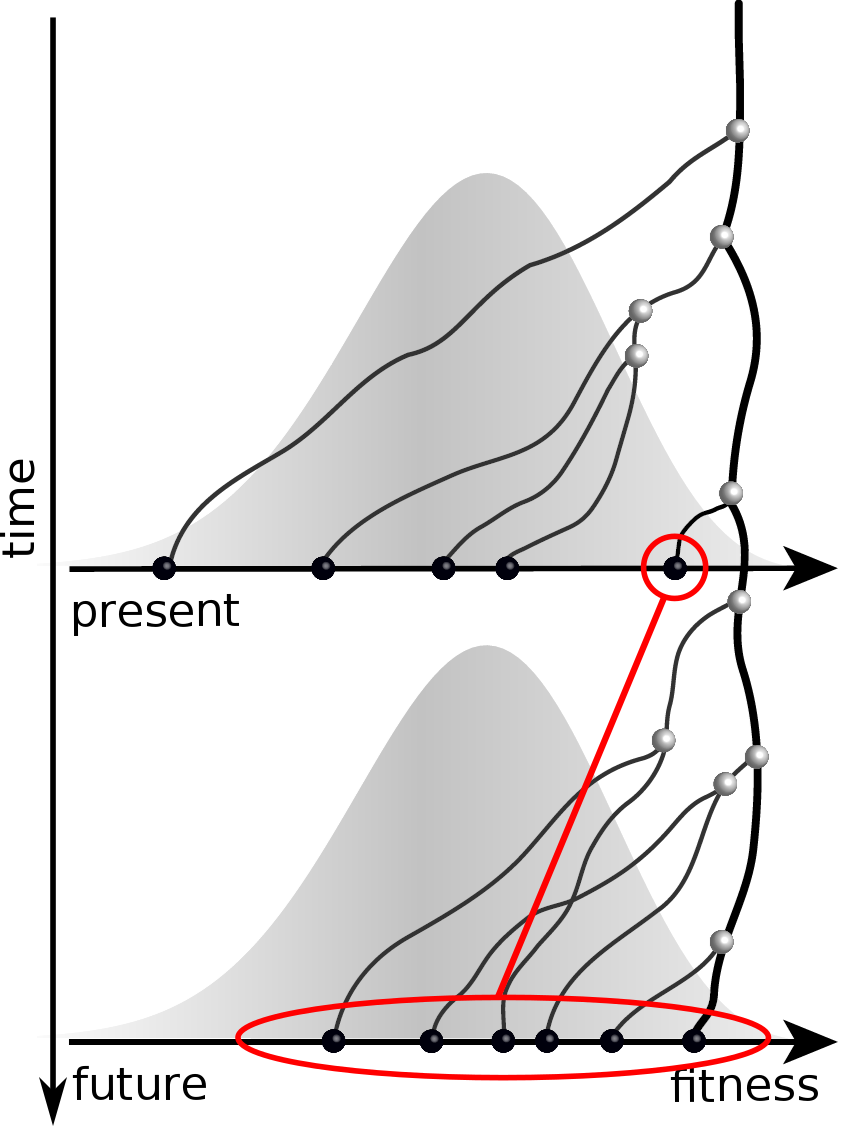
Predicting evolution
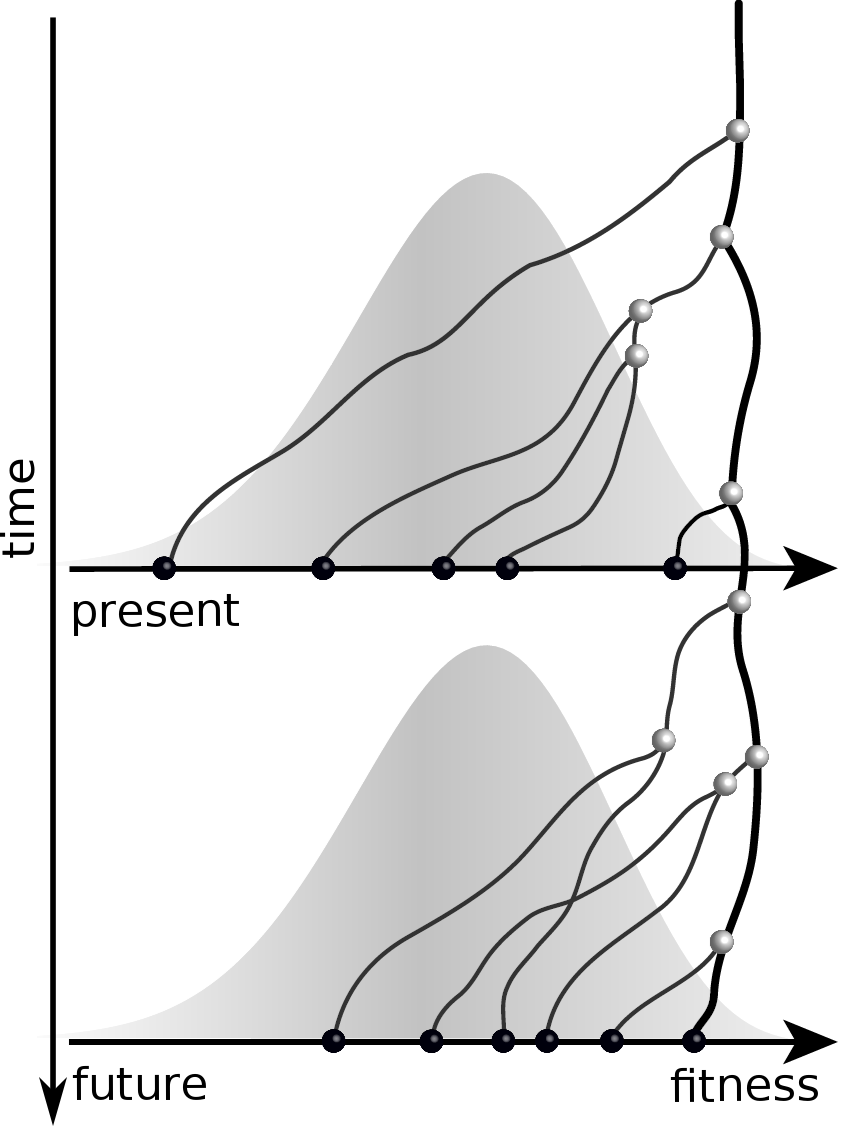
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
Validate on simulation data
- simulate evolution
- sample sequences
- reconstruct trees
- infer fitness
- predict ancestor of future
- compare to truth
Validation on simulated data
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
Hemagglutination Inhibition assays
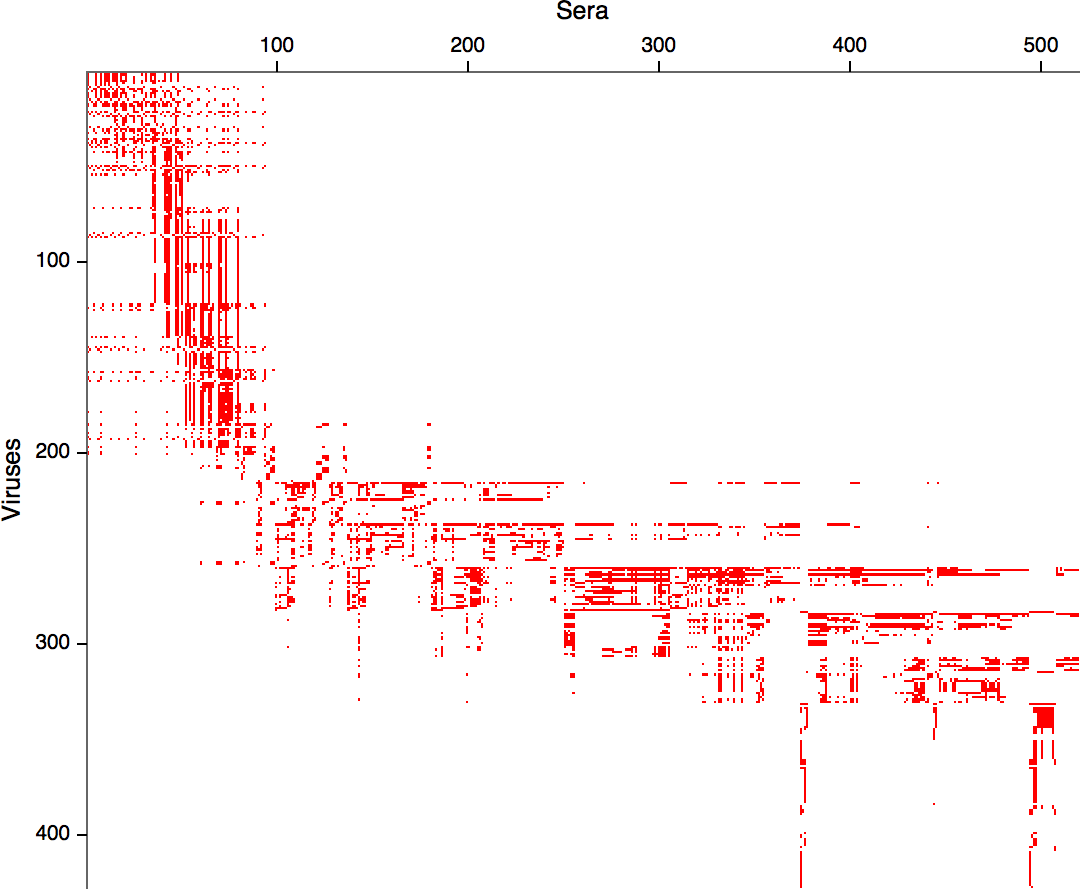
HI data sets
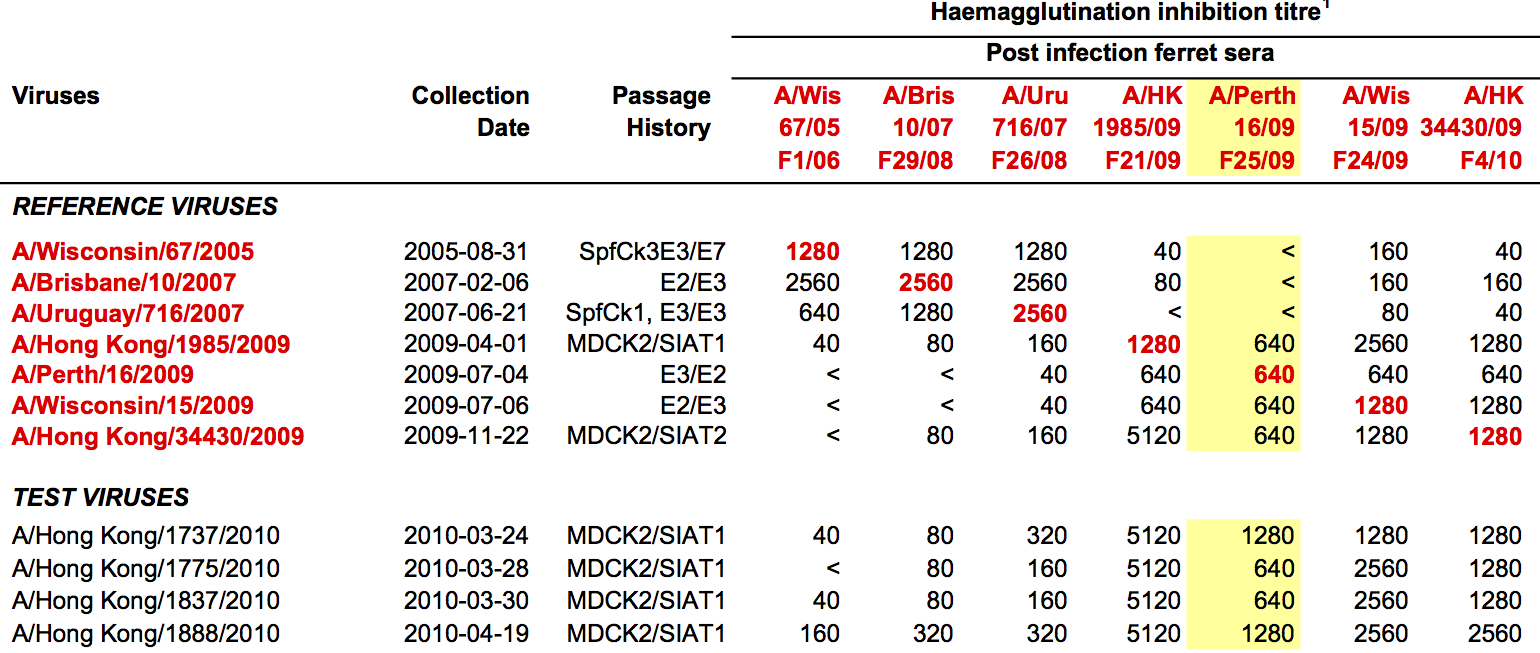
- Long list of distances between sera and viruses
- Tables are sparse, only close by pairs
- Structure of space is not immediately clear
- MDS in 2 or 3 dimensions
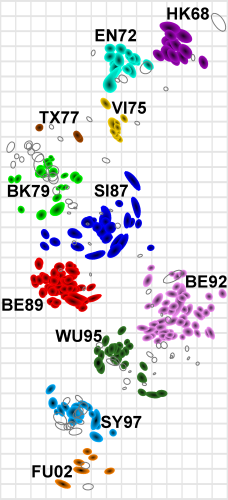 Smith et al, Science 2002
Smith et al, Science 2002
Integrating antigenic and molecular evolution

- $H_{a\beta} = v_a + p_\beta + \sum_{i\in (a,b)} d_i$
- each branch contributes $d_i$ to antigenic distance
- sparse solution for $d_i$ through $l_1$ regularization
- related model where $d_i$ are associated with substitutions
Integrating antigenic and molecular evolution

- MDS: $(d+1)$ parameters per virus
- Tree model: $2$ parameters per virus
- Sparse solution
→ identify branches or substitutions that cause titer drop
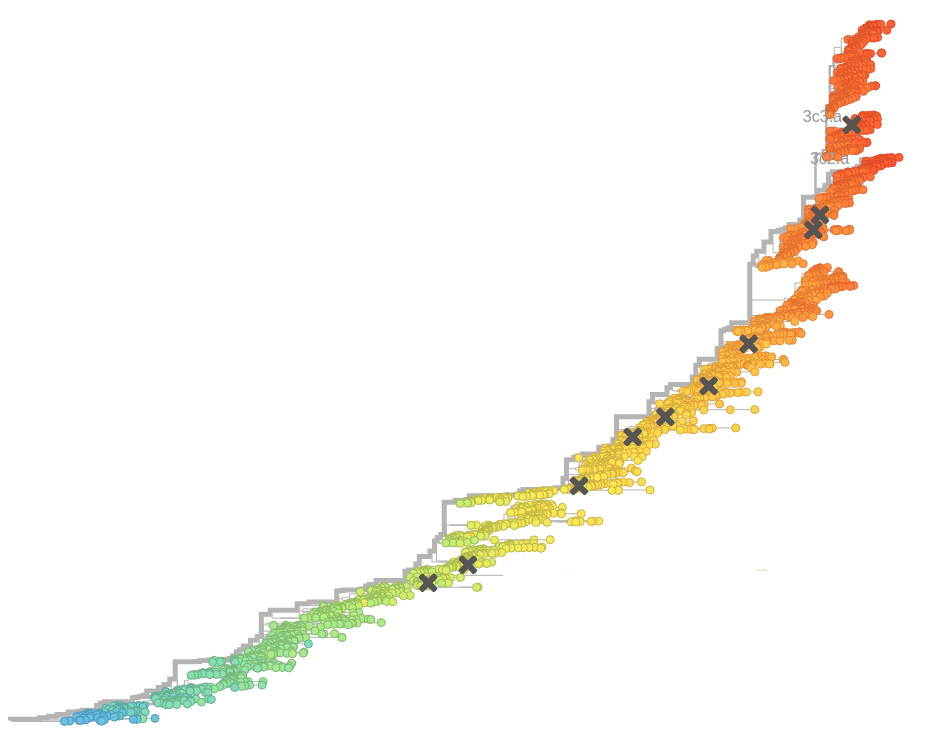
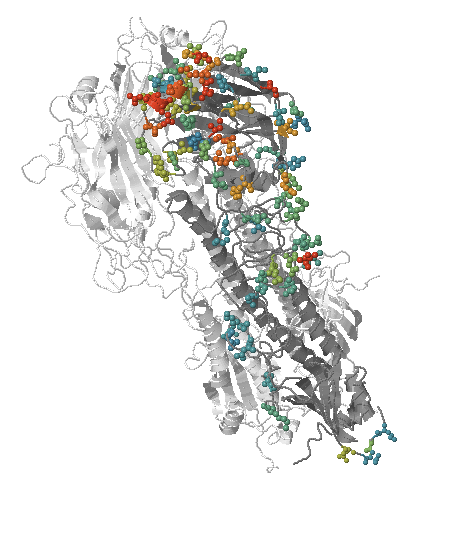
HI distances on the phylogenetic tree
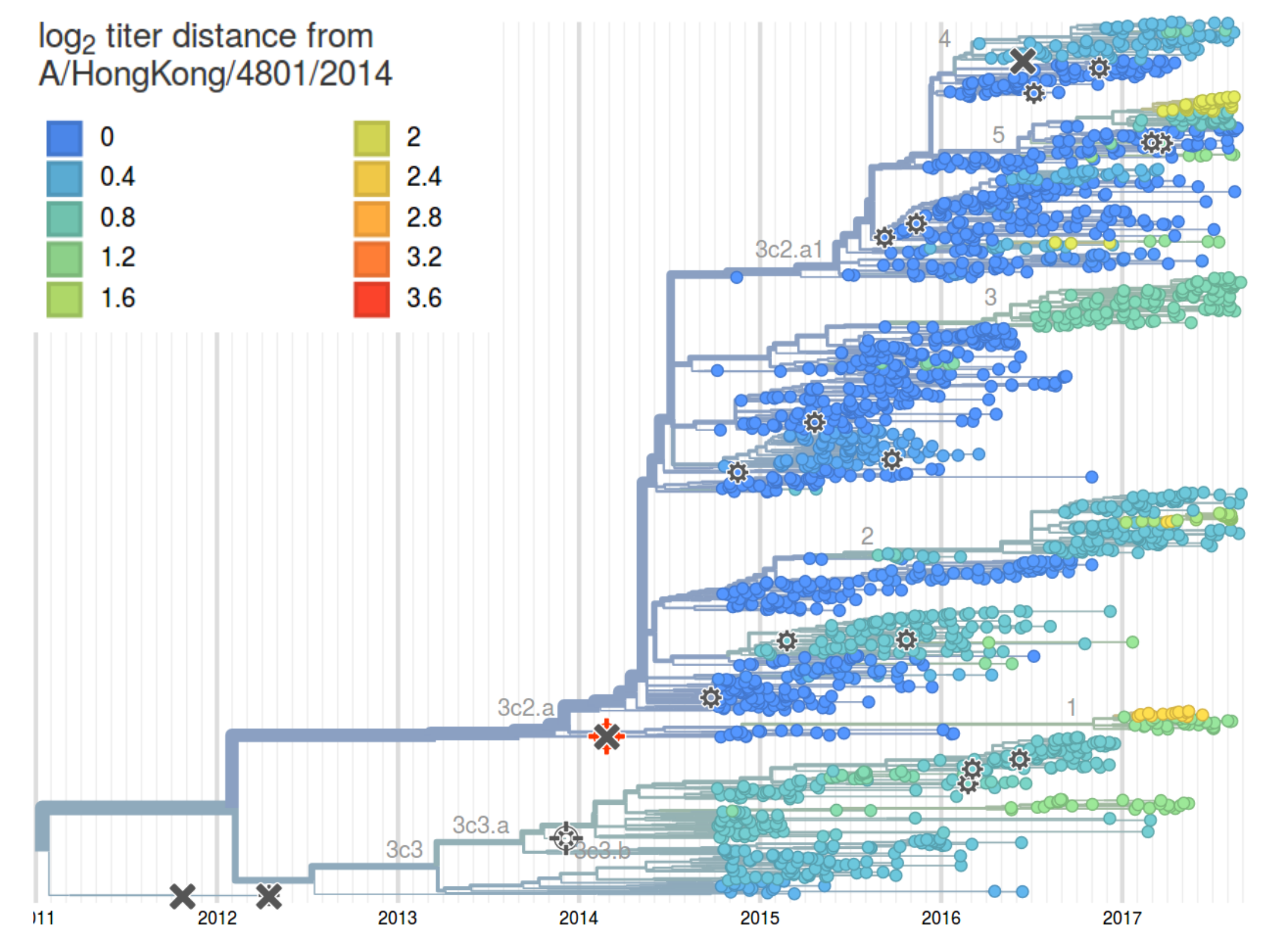
nextstrain.org
joint work with Trevor Bedford & his lab
NextStrain architecture
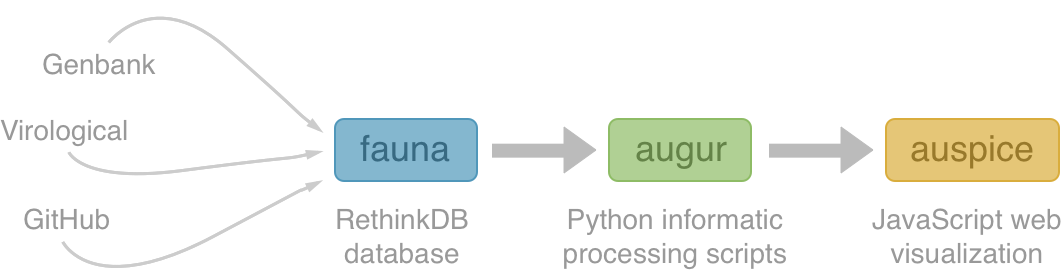
Using treetime to rapidly compute timetrees
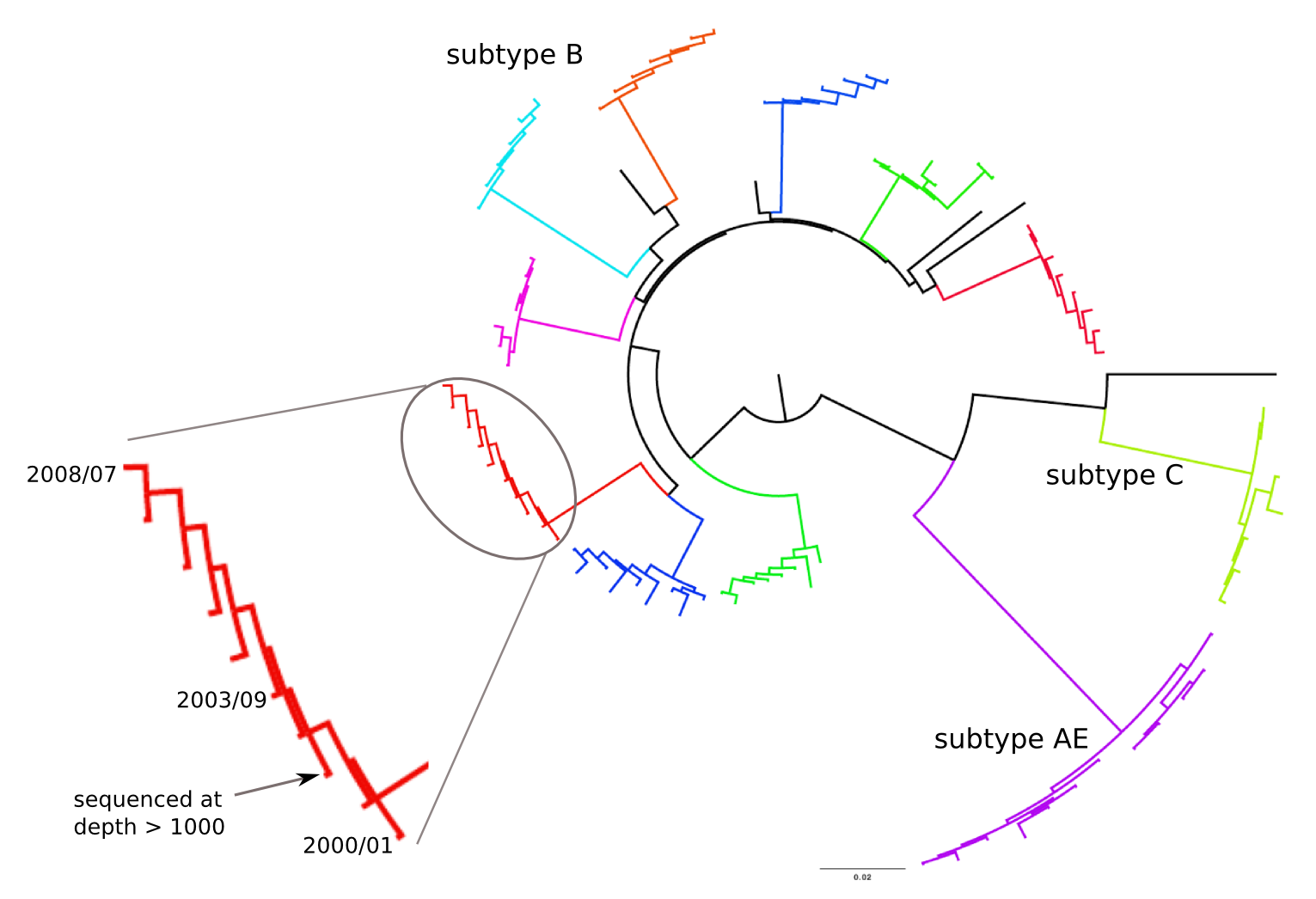
HIV-1 evolution within one individual

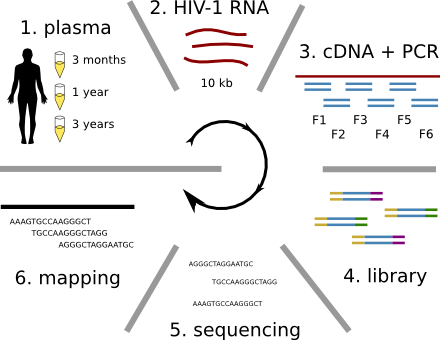

HIV-1 sequencing before and after therapy
 Zanini et al, eLife, 2015;
Brodin et al, eLife, 2016.
Collaboration with the group of Jan Albert
Zanini et al, eLife, 2015;
Brodin et al, eLife, 2016.
Collaboration with the group of Jan Albert
Inference of fitness costs
- mutation away from preferred state with rate $\mu$
- selection against non-preferred state with strength $s$
- variant frequency dynamics: $\frac{d x}{dt} = \mu -s x $
- equilibrium frequency: $\bar{x} = \mu/s $
- fitness cost: $s = \mu/\bar{x}$
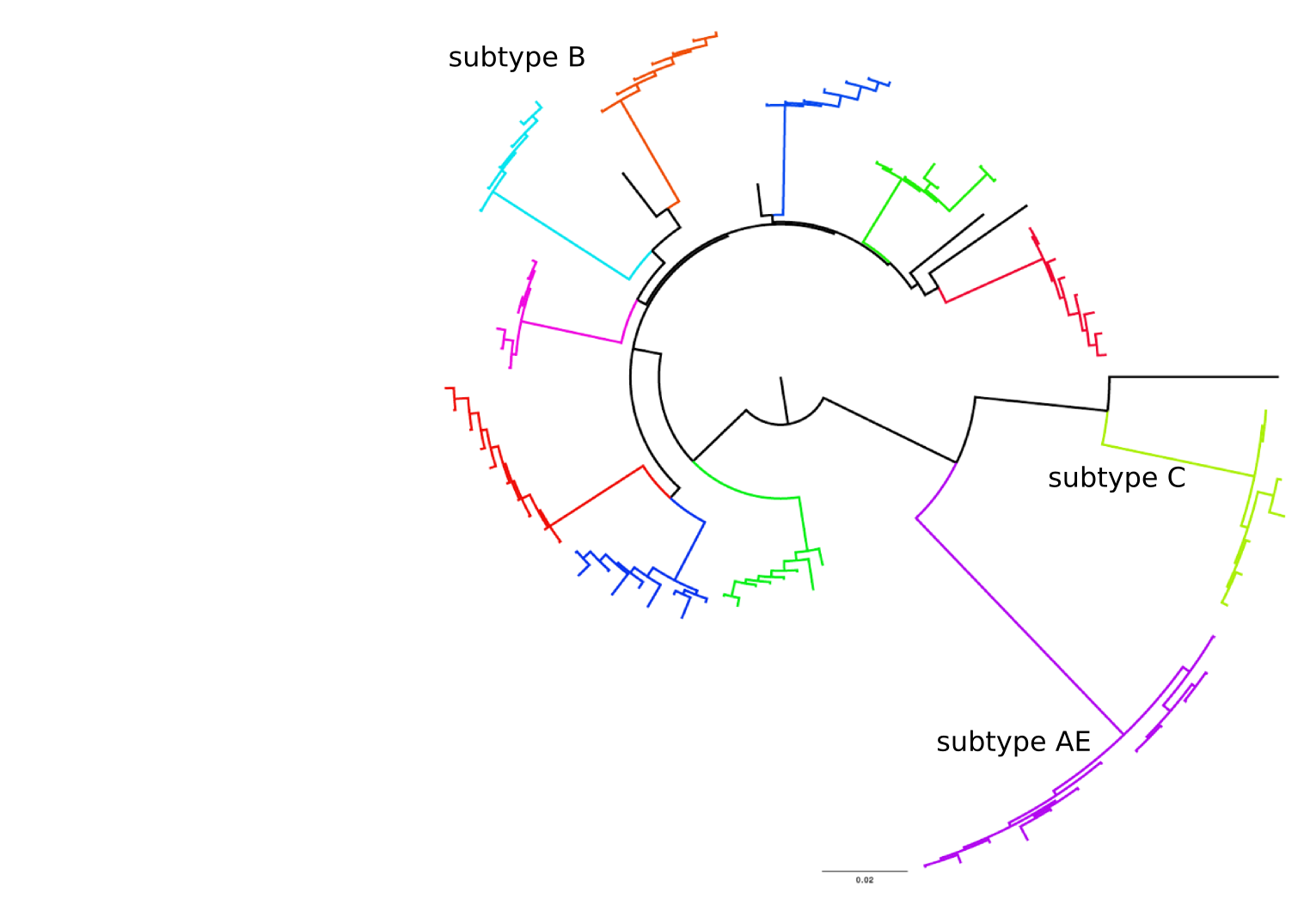
Fitness landscape of HIV-1
Zanini et al, Virus Evolution, 2017Selection on RNA structures and regulatory sites
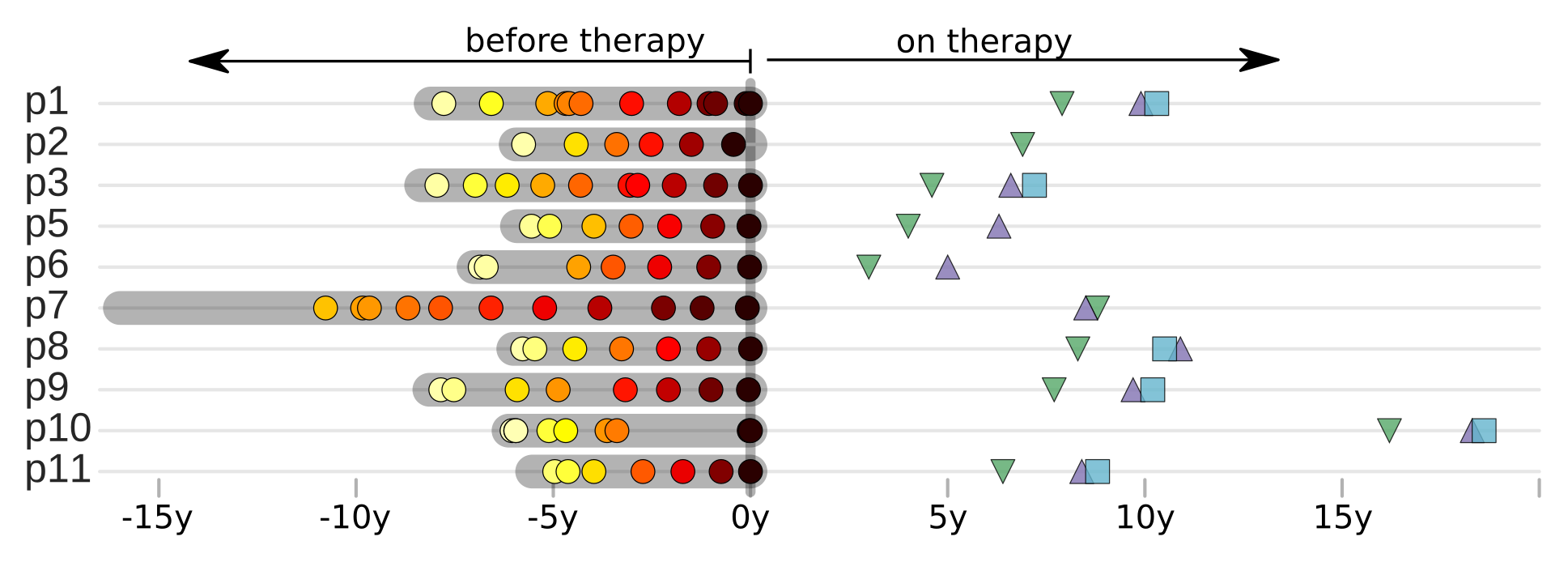
Zanini et al, Virus Evolution, 2017Does HIV evolve during therapy?
Brodin et al, eLife, 2016
No evidence of ongoing replication
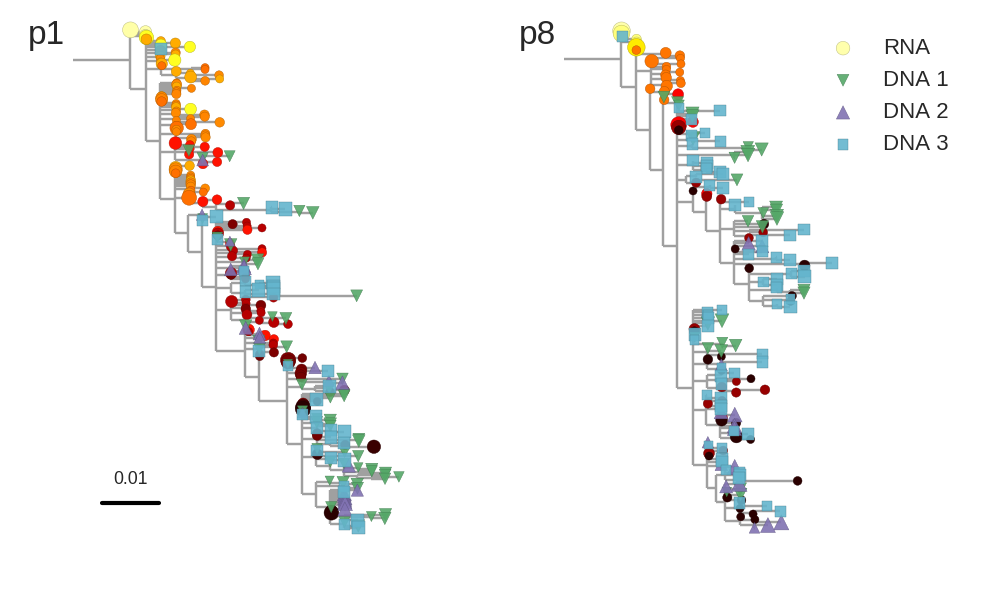
No evidence of ongoing replication
 Brodin et al, eLife, 2016
Brodin et al, eLife, 2016
T-cell turnover is fast in untreated infection

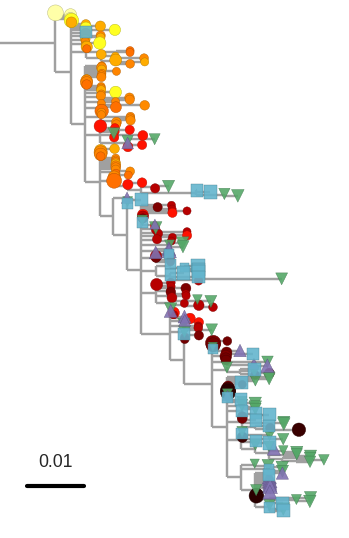
- latent HIV → barcode of a T-cell lineage
- all latent integrated virus derives from late infection
- untreated: T-cell lineages are short lived
- on therapy: T-cell clones live decades
Summary
- RNA virus evolution can be observed directly
- Rapidly adapting population require new population genetic models
- Those model can be used to infer fit clades
- Future influenza population can be anticipated
- Automated real-time analysis can help fight the spread of disease
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
nextstrain.org





- Trevor Bedford
- Colin Megill
- Pavel Sagulenko
- Sidney Bell
- James Hadfield
- Wei Ding

Acknowledgments
- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller