Applied evolutionary biology: tracking and predicting the spread of disease
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201712_ICTP2.html
Sequences record the spread of pathogens
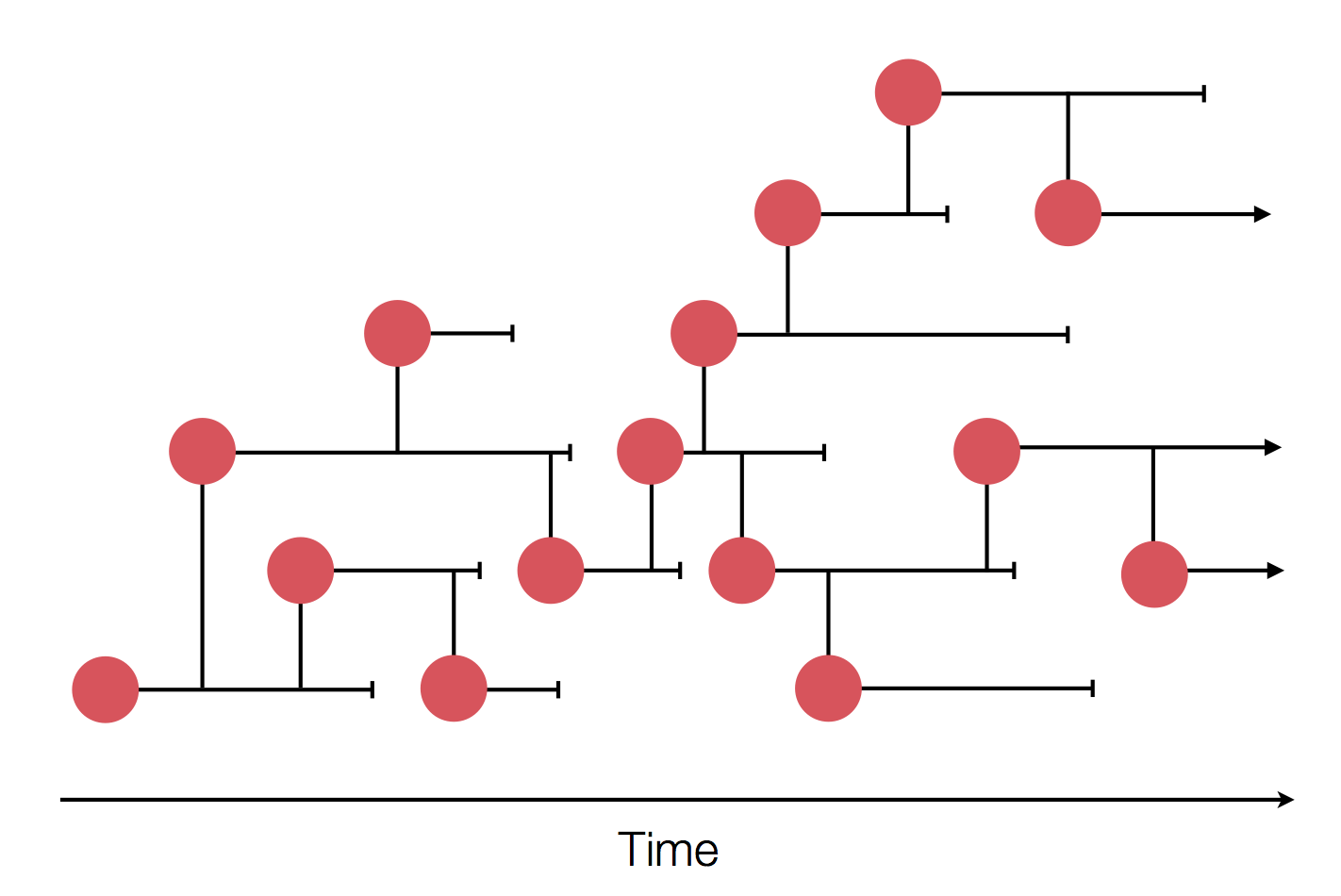
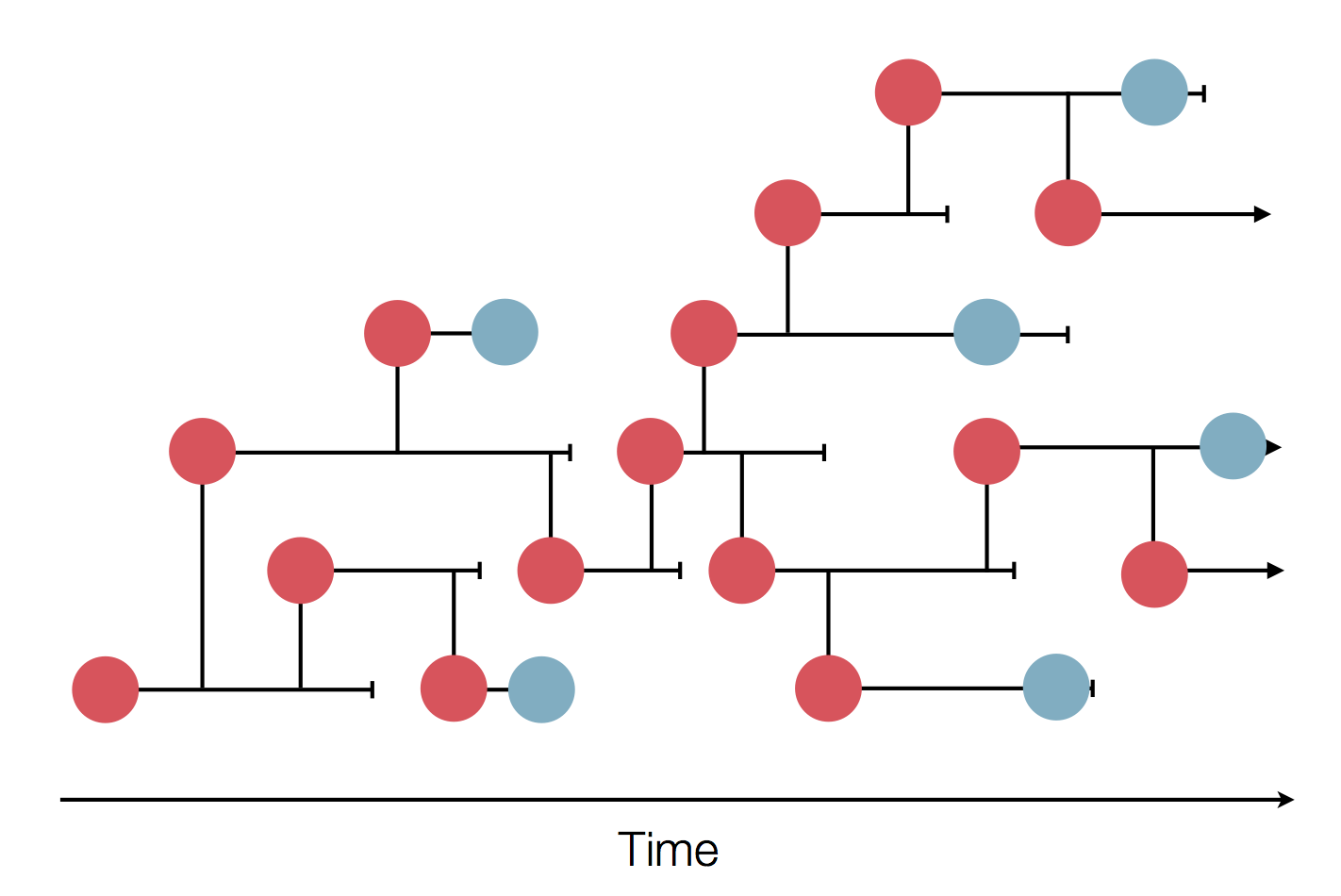
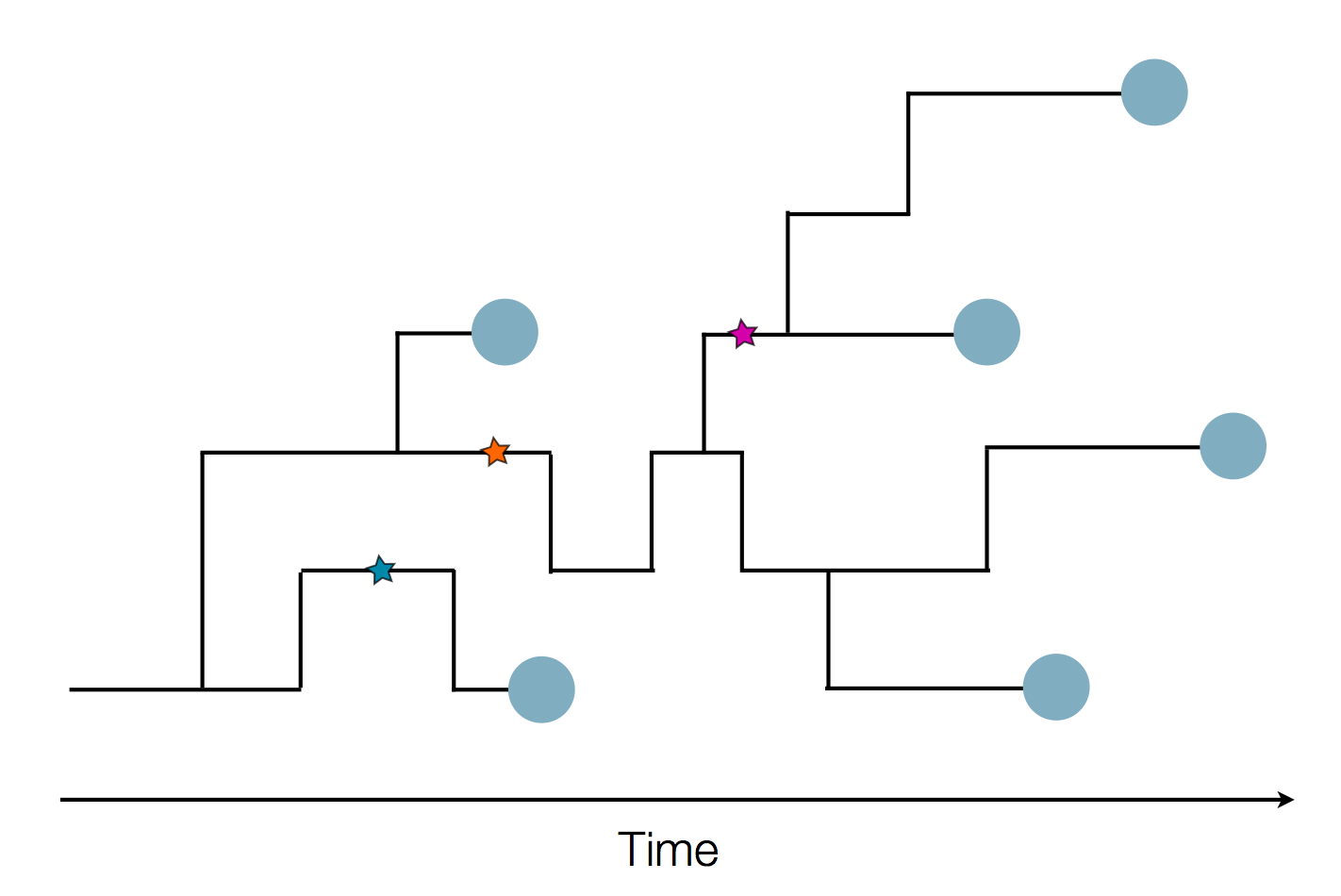
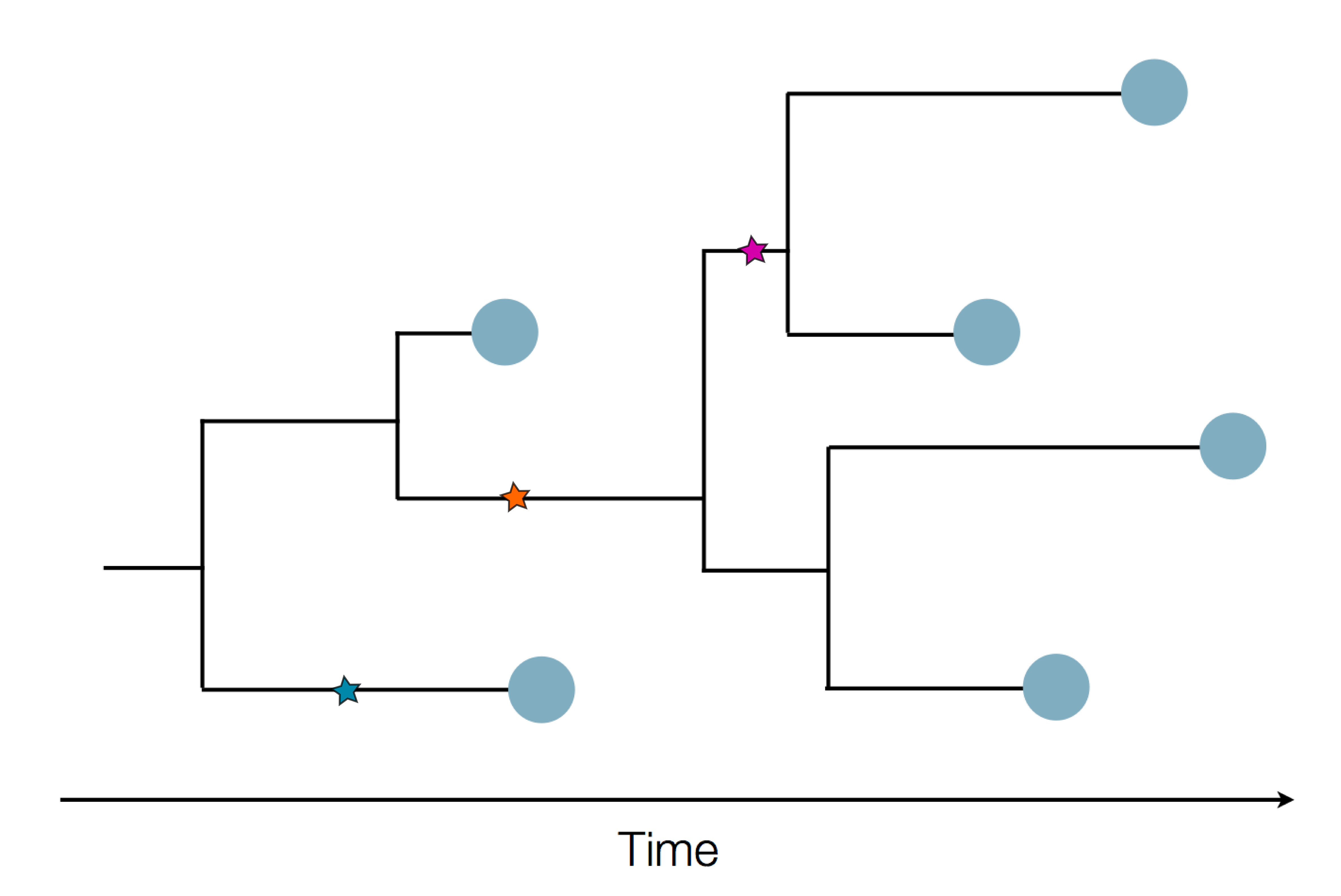
The resolution is limited by the number of mutations!
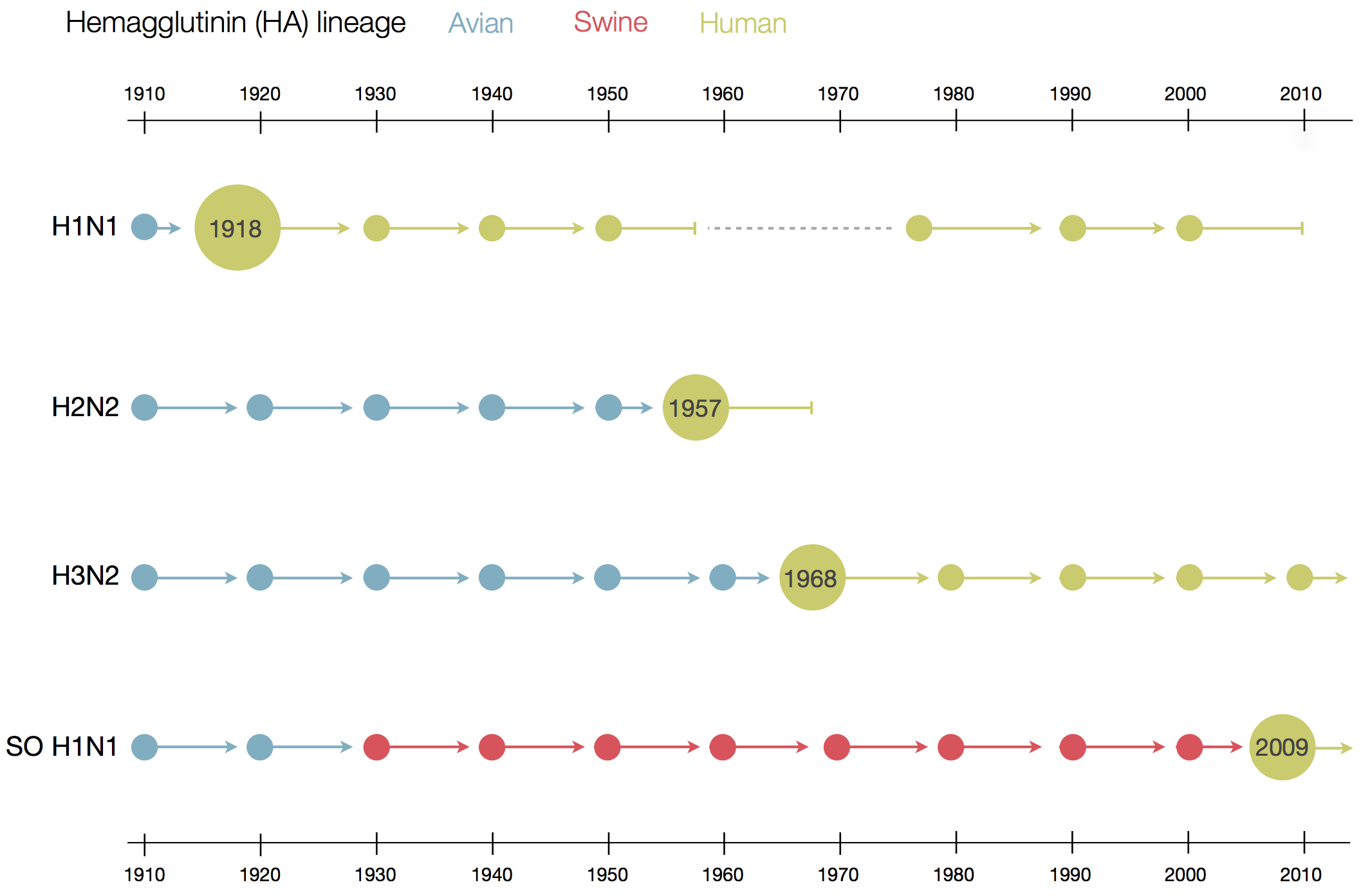
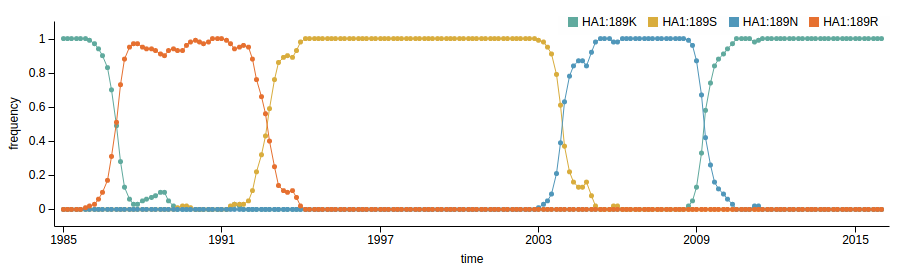
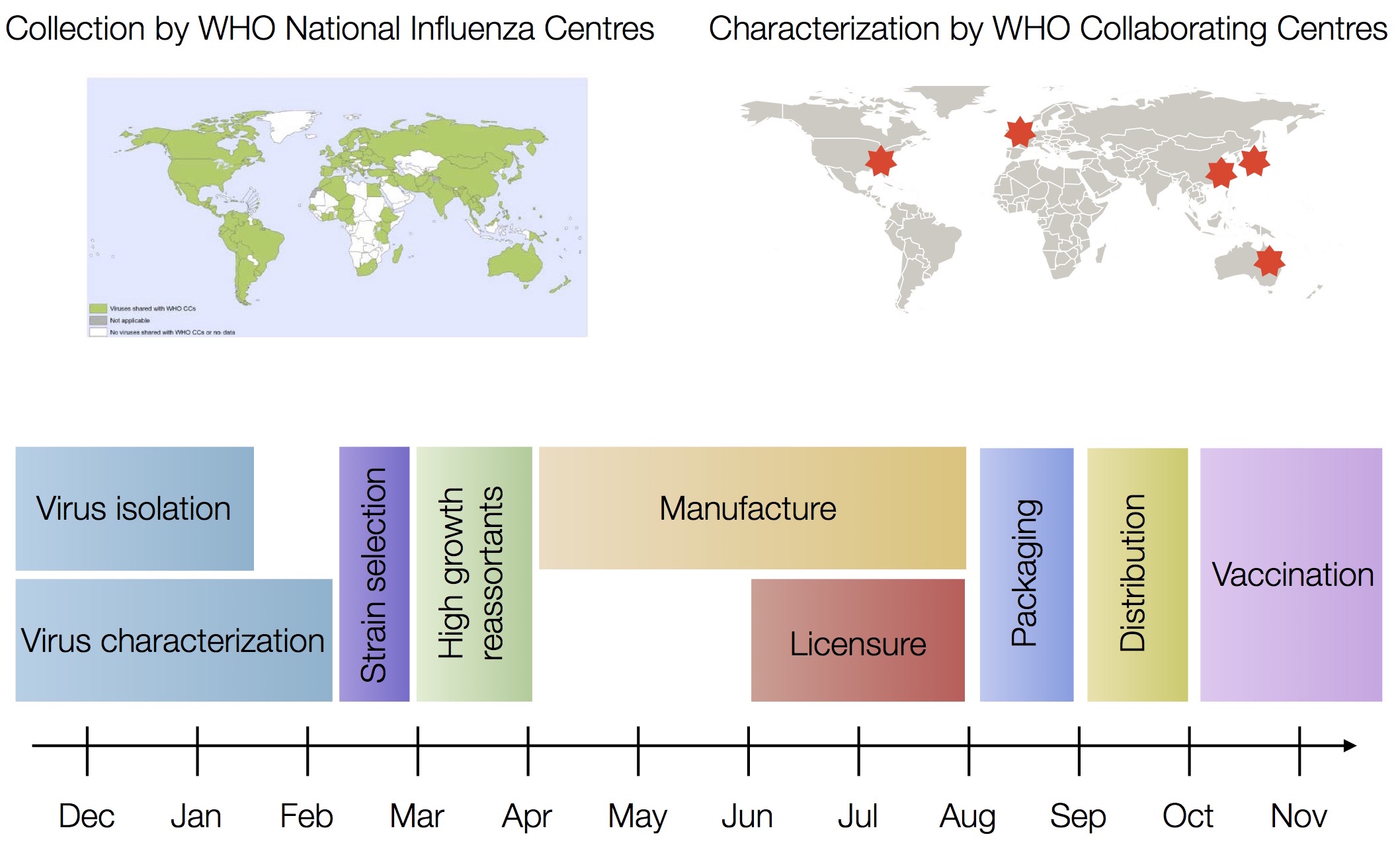
Human seasonal influenza viruses
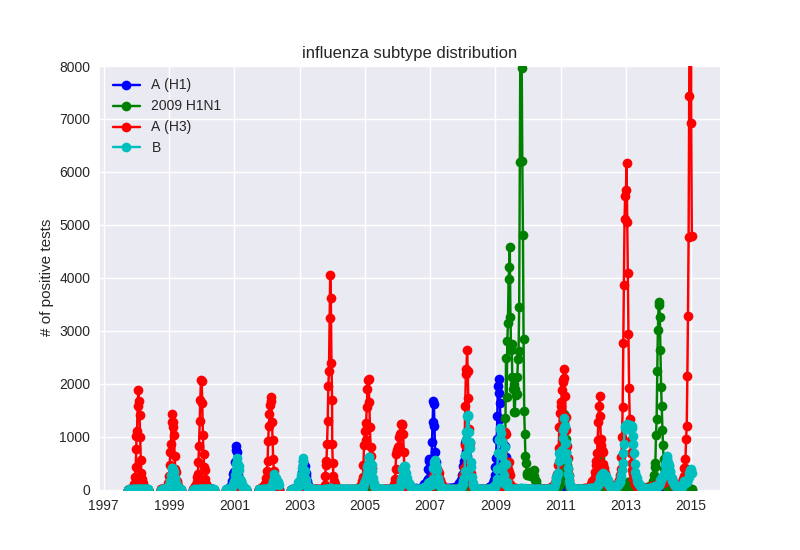
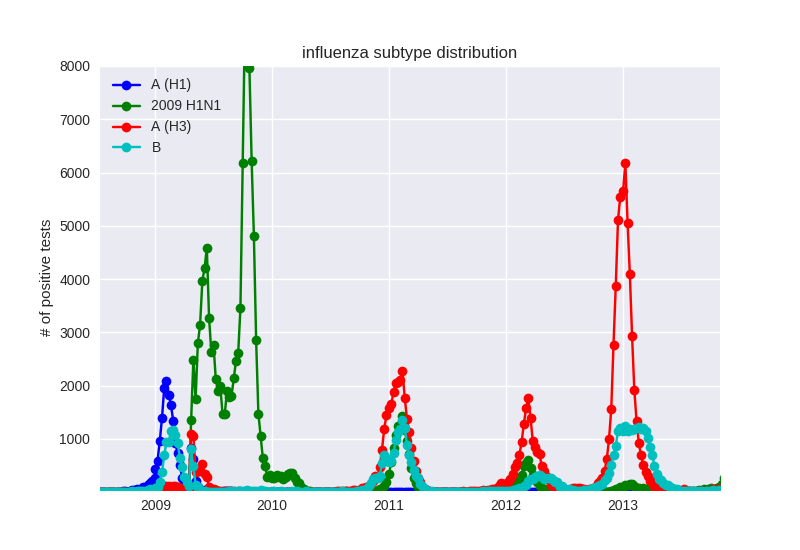
Influenza seasonality - USA
- Influenza viruses evolve to avoid human immunity
- Vaccines need frequent updates
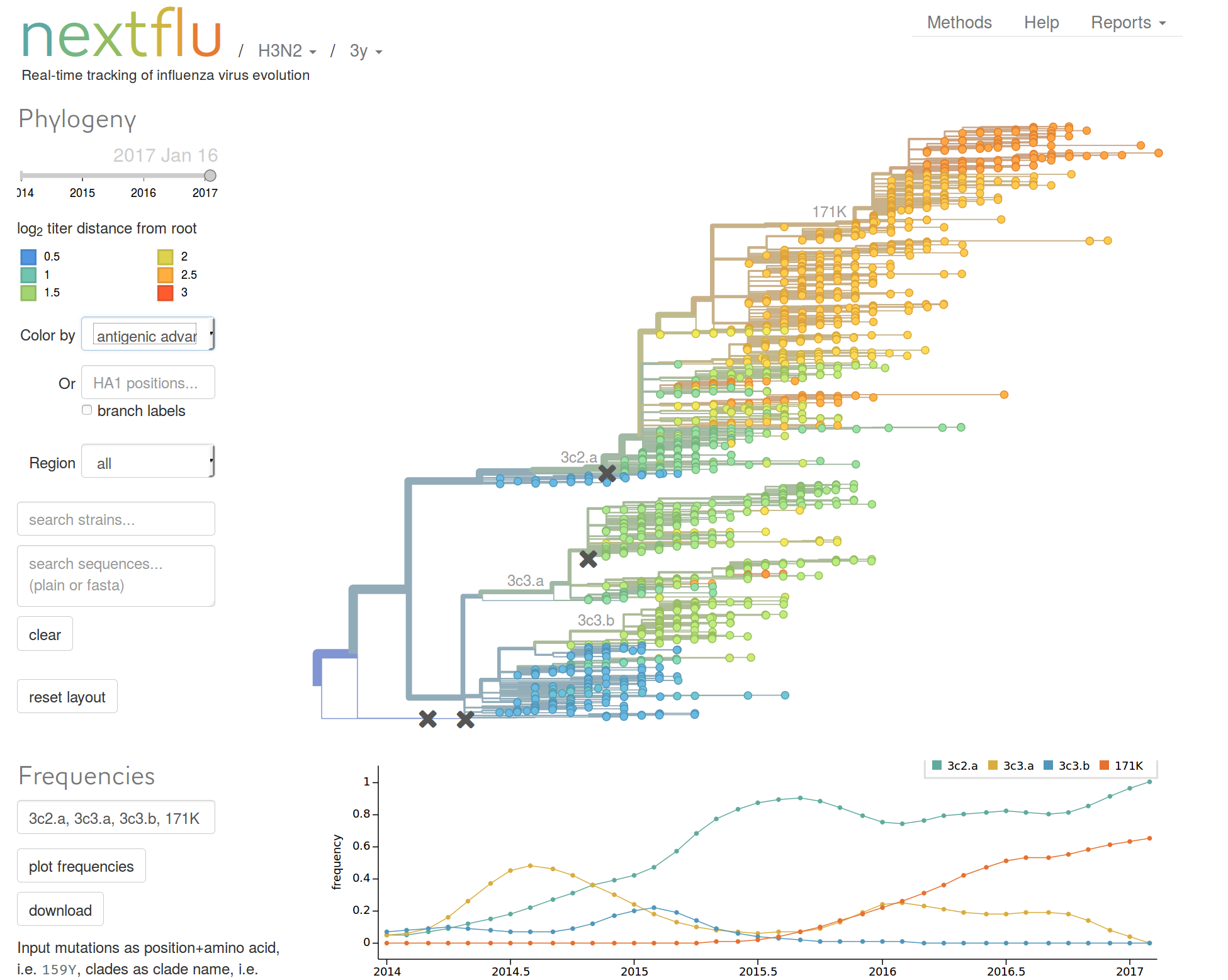
nextflu.org
joint work with Trevor Bedford & his lab
Beyond tracking: can we predict?
 slide by Trevor Bedford
slide by Trevor Bedford
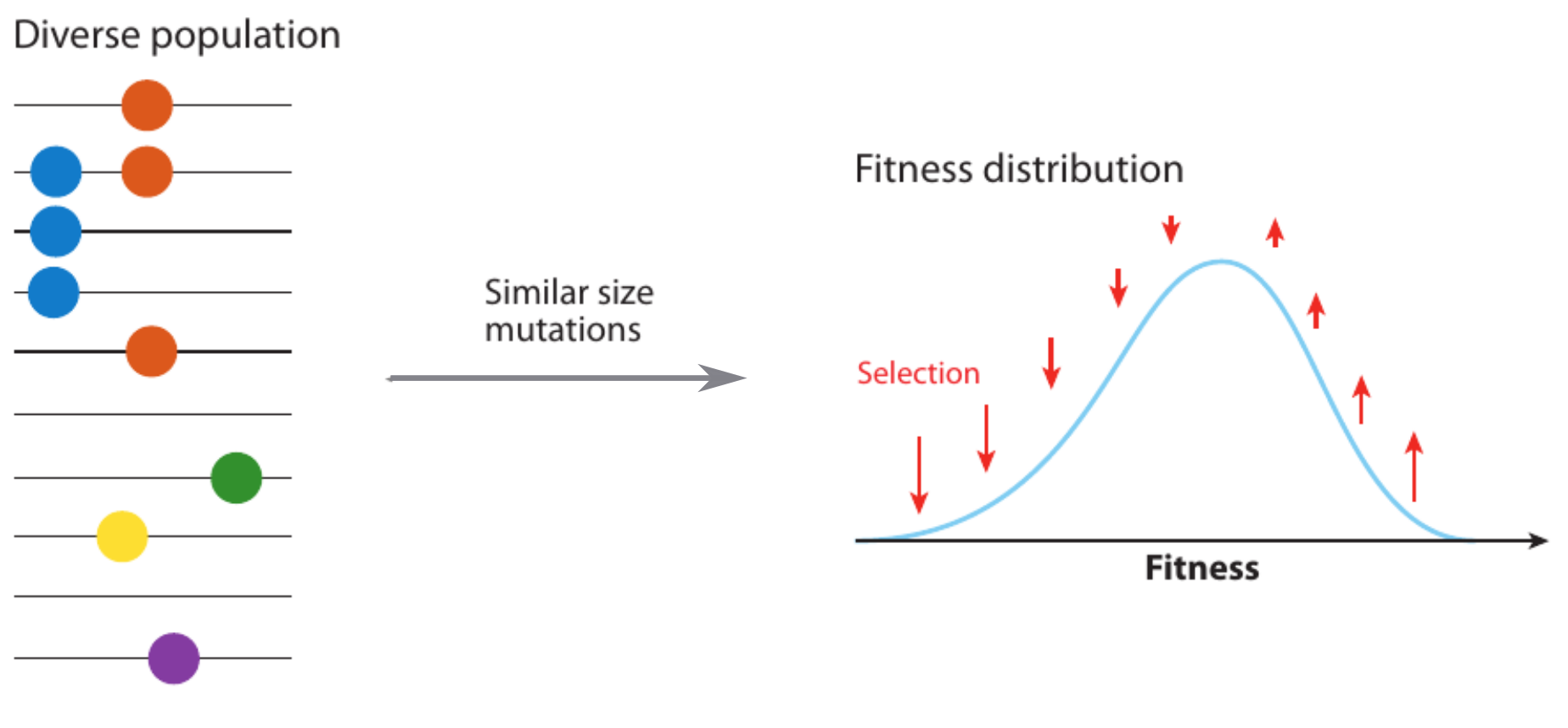
Clonal interference and traveling waves
Influenza virus prediction by Luksza & Lässig
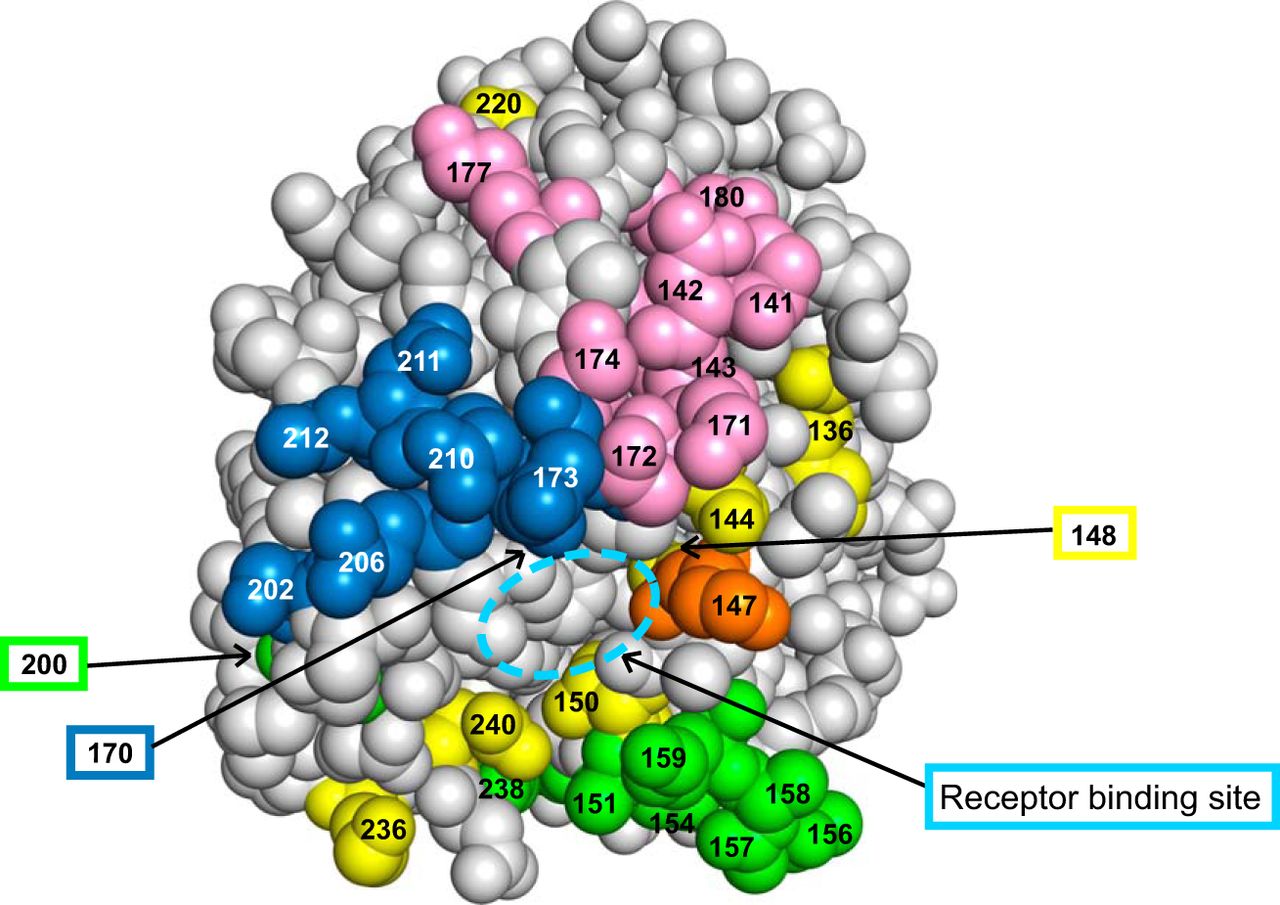
- Epitope mutations: association with antigenic change
- Non-epitope mutations: likely deleterious
- Nonlinear component: synonymous mutations
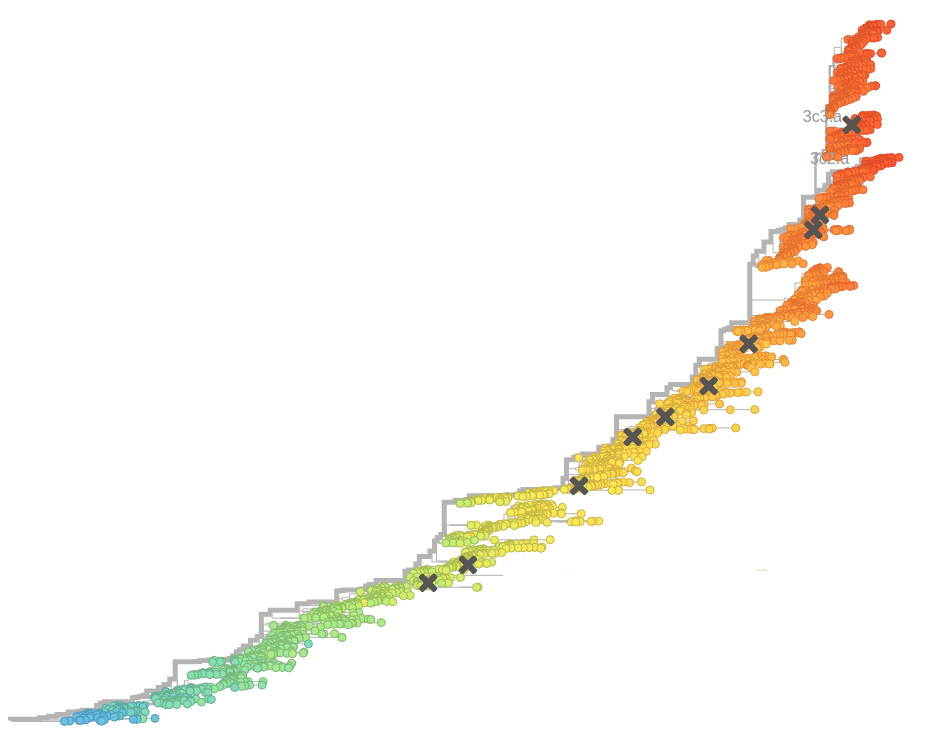
Typical tree
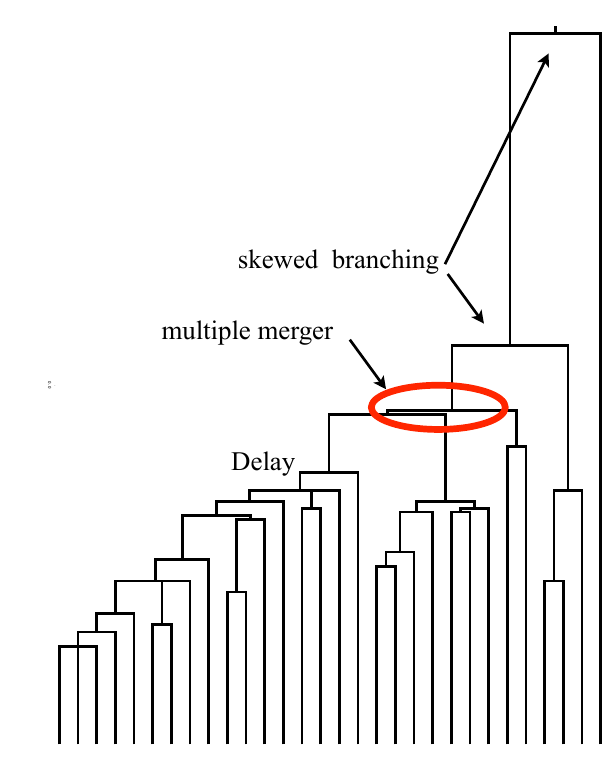
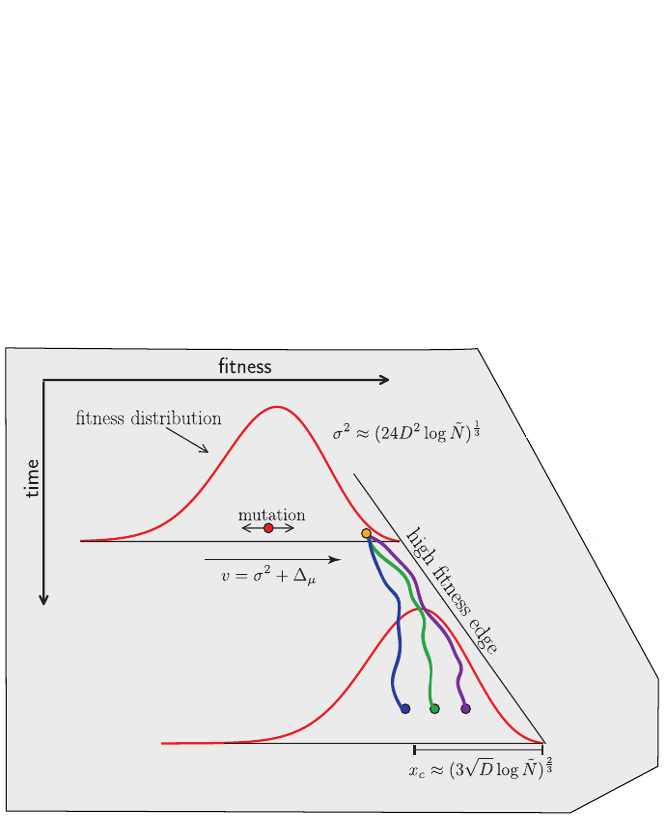
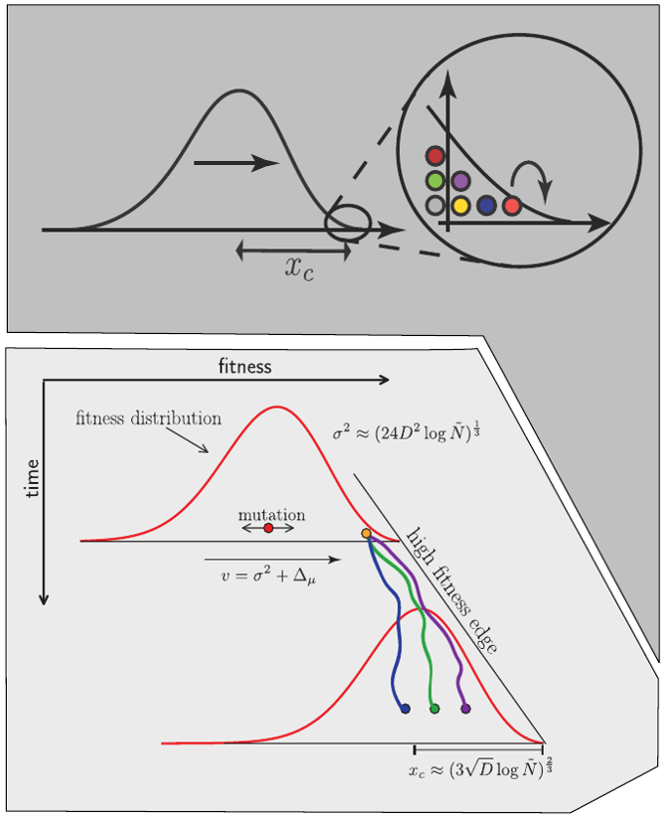
Bolthausen-Sznitman Coalescent
Predicting evolution
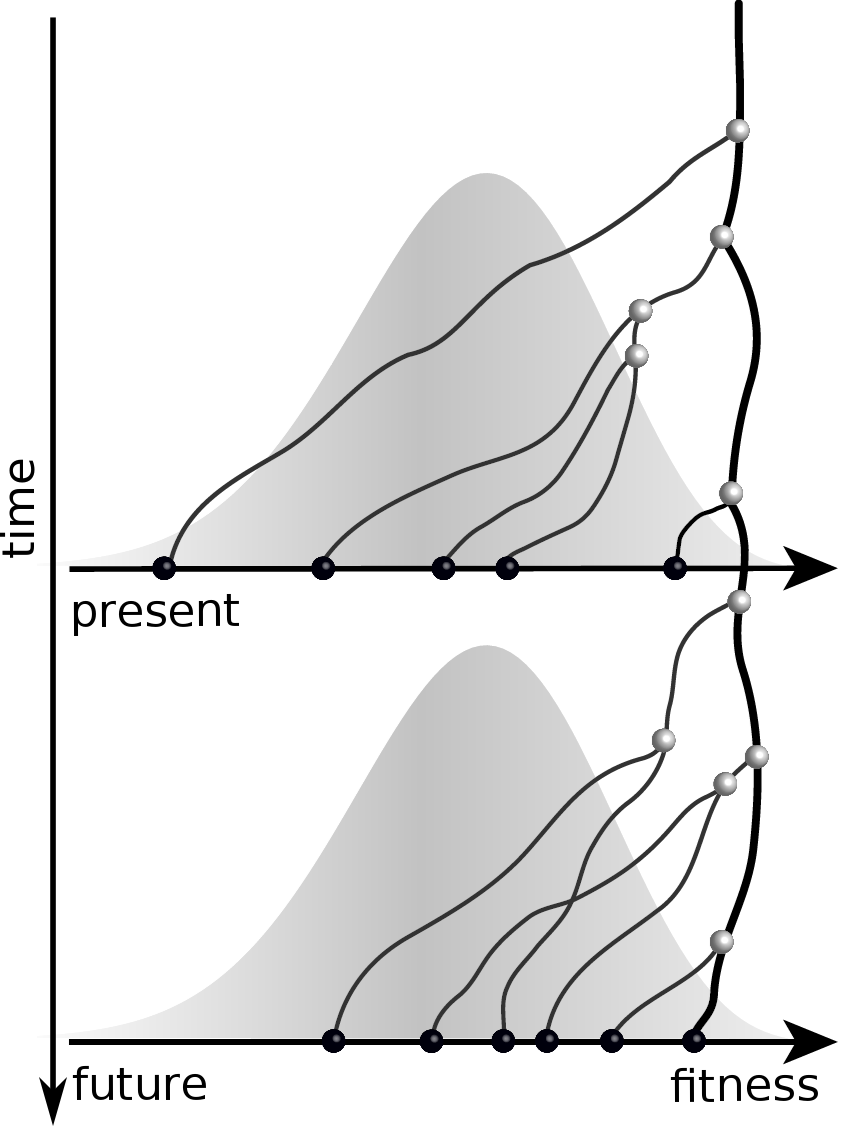
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
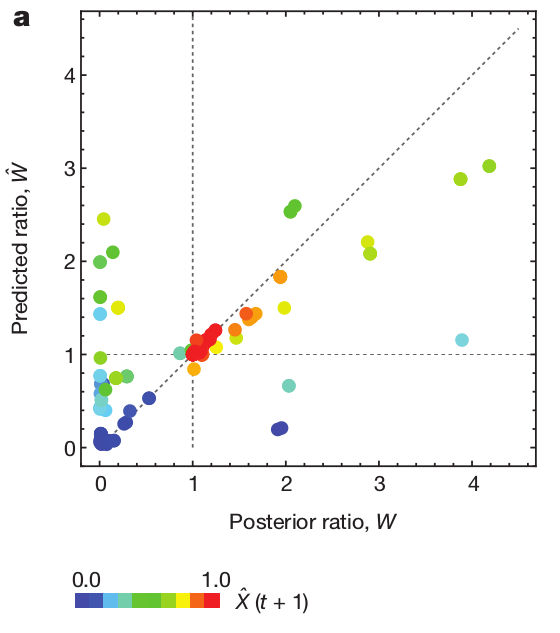
Validation on simulated data
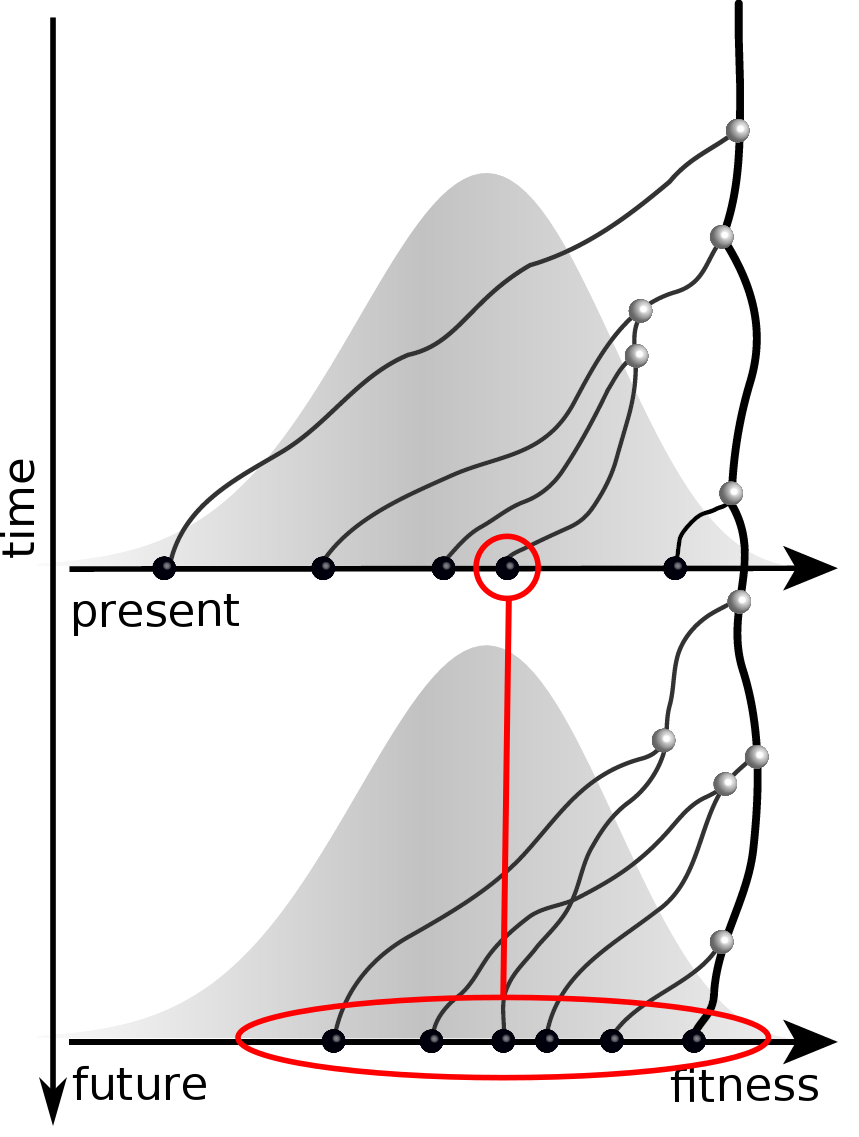
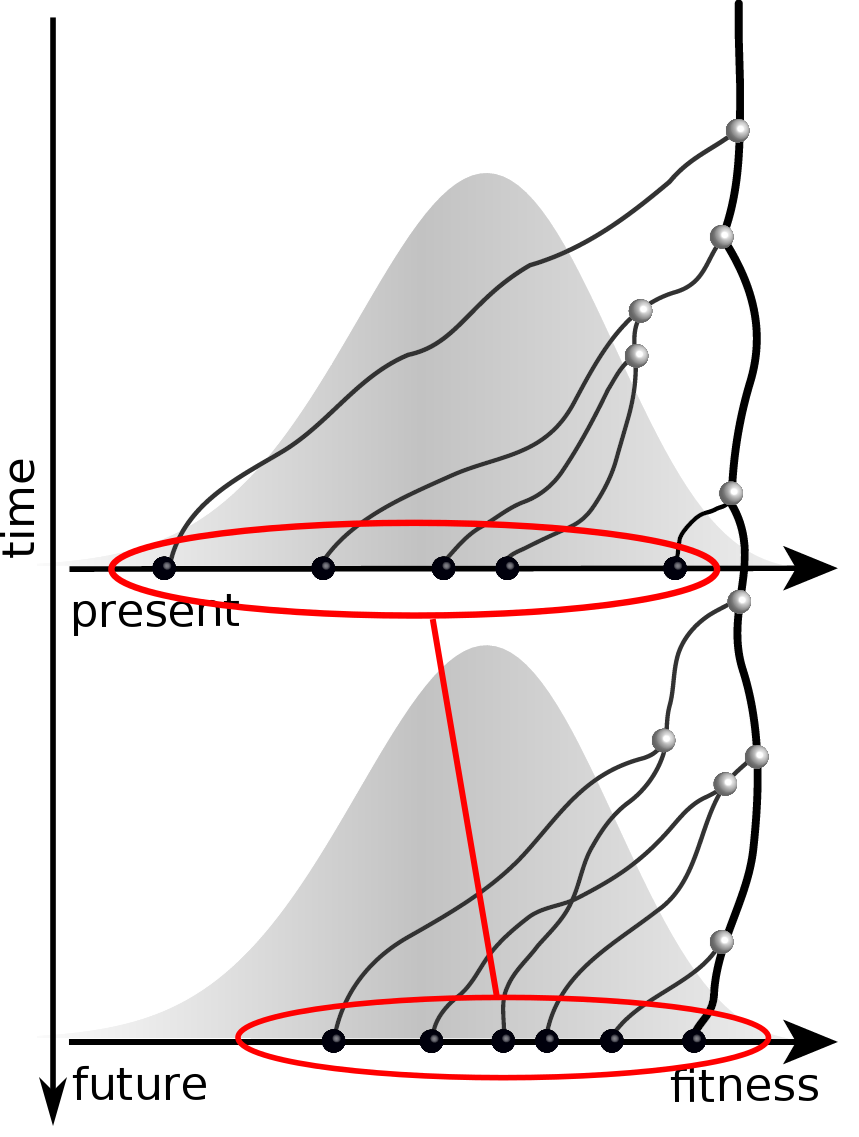
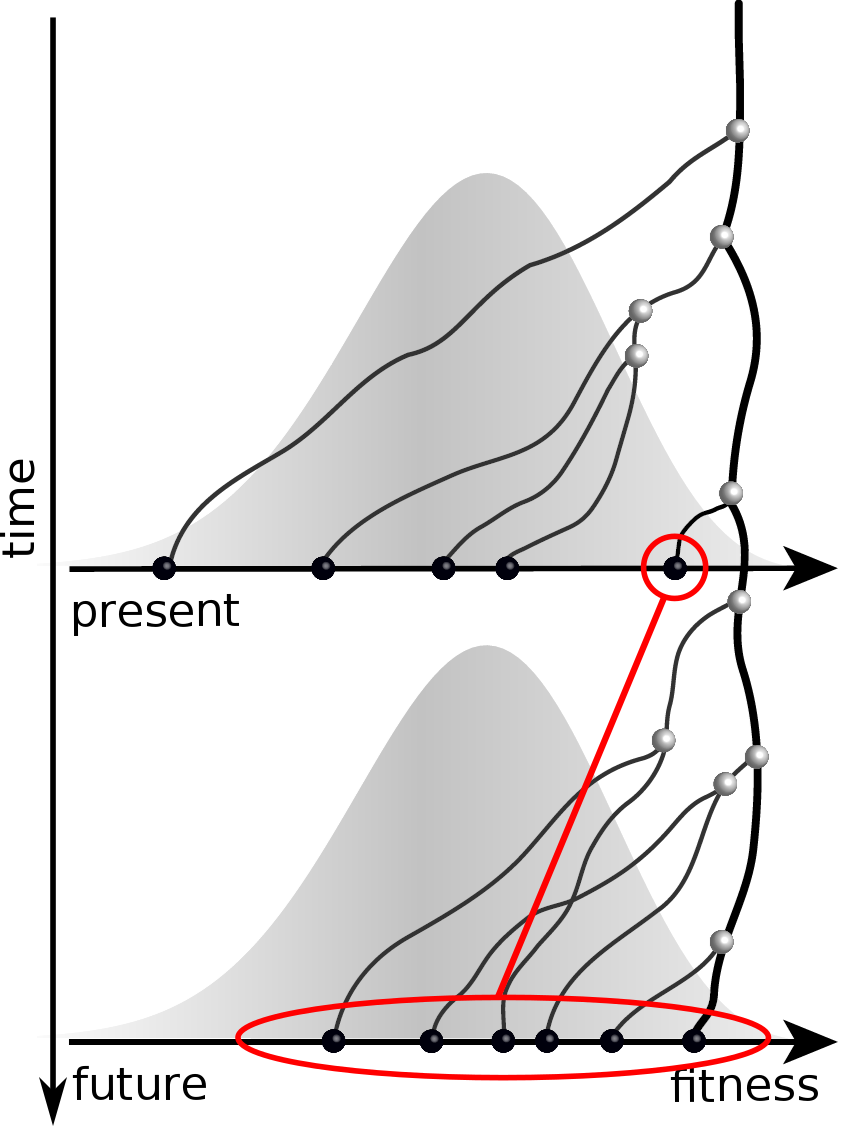
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
Hemagglutination Inhibition assays
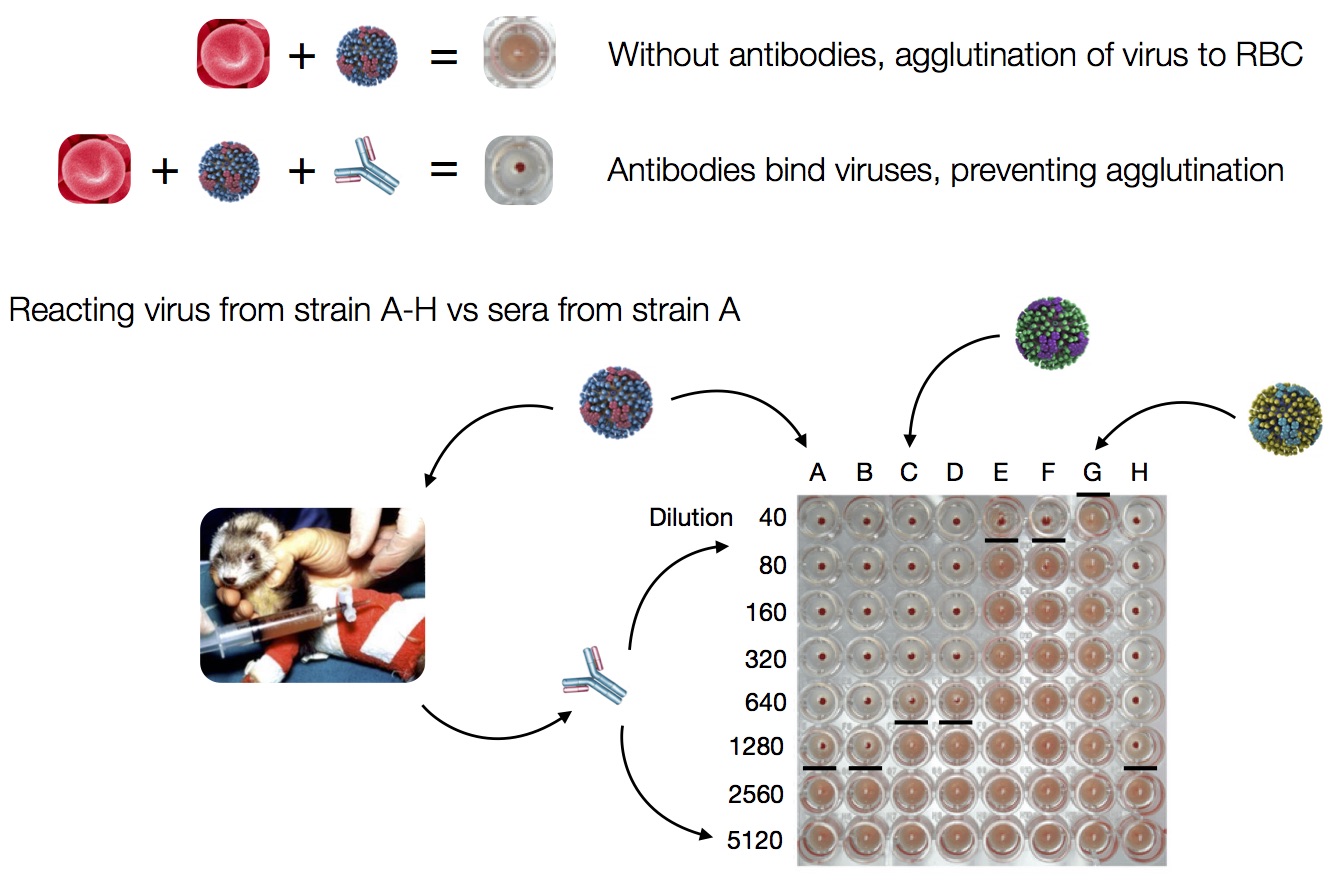
HI data sets
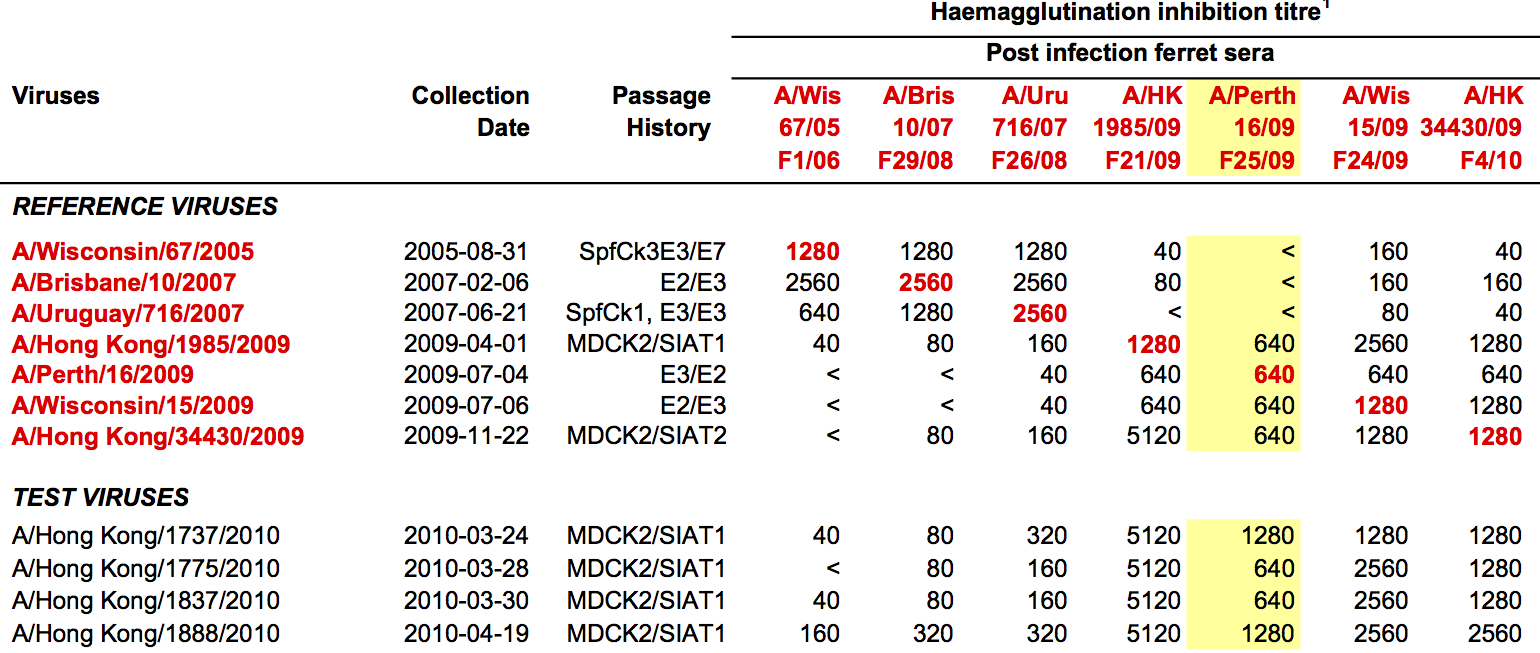
- Long list of distances between sera and viruses
- Tables are sparse, only close by pairs
- Structure of space is not immediately clear
- MDS in 2 or 3 dimensions
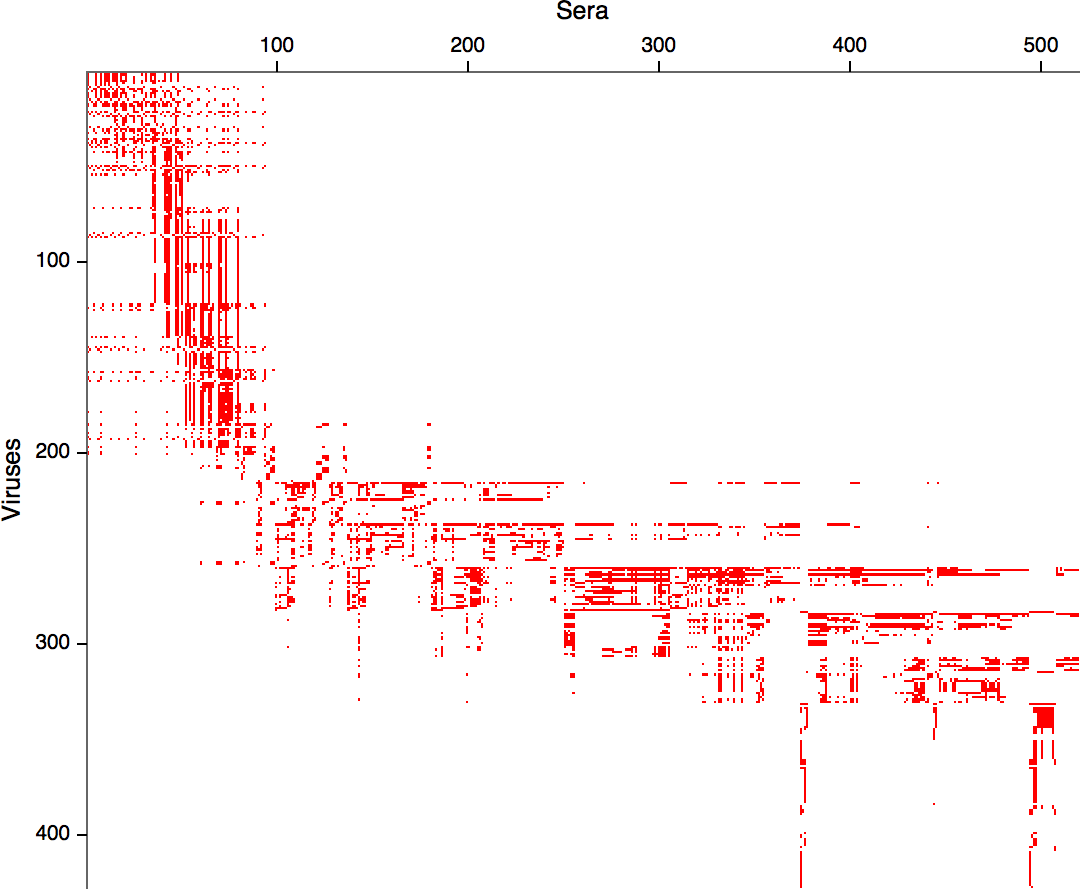
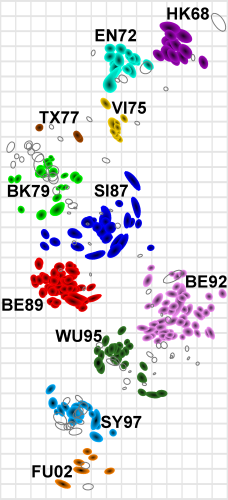 Smith et al, Science 2002
Smith et al, Science 2002
Integrating antigenic and molecular evolution

- $H_{a\beta} = v_a + p_\beta + \sum_{i\in (a,b)} d_i$
- each branch contributes $d_i$ to antigenic distance
- sparse solution for $d_i$ through $l_1$ regularization
- related model where $d_i$ are associated with substitutions
Integrating antigenic and molecular evolution

- MDS: $(d+1)$ parameters per virus
- Tree model: $2$ parameters per virus
- Sparse solution
→ identify branches or substitutions that cause titer drop
Are antigenic distances tree-like?
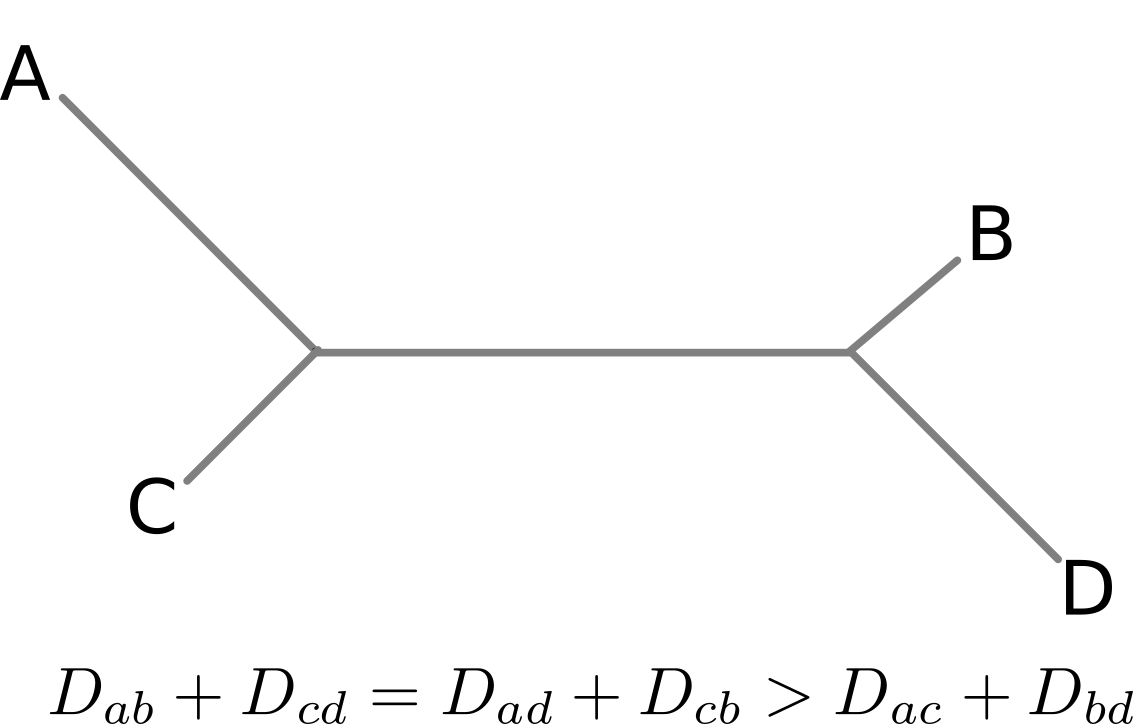
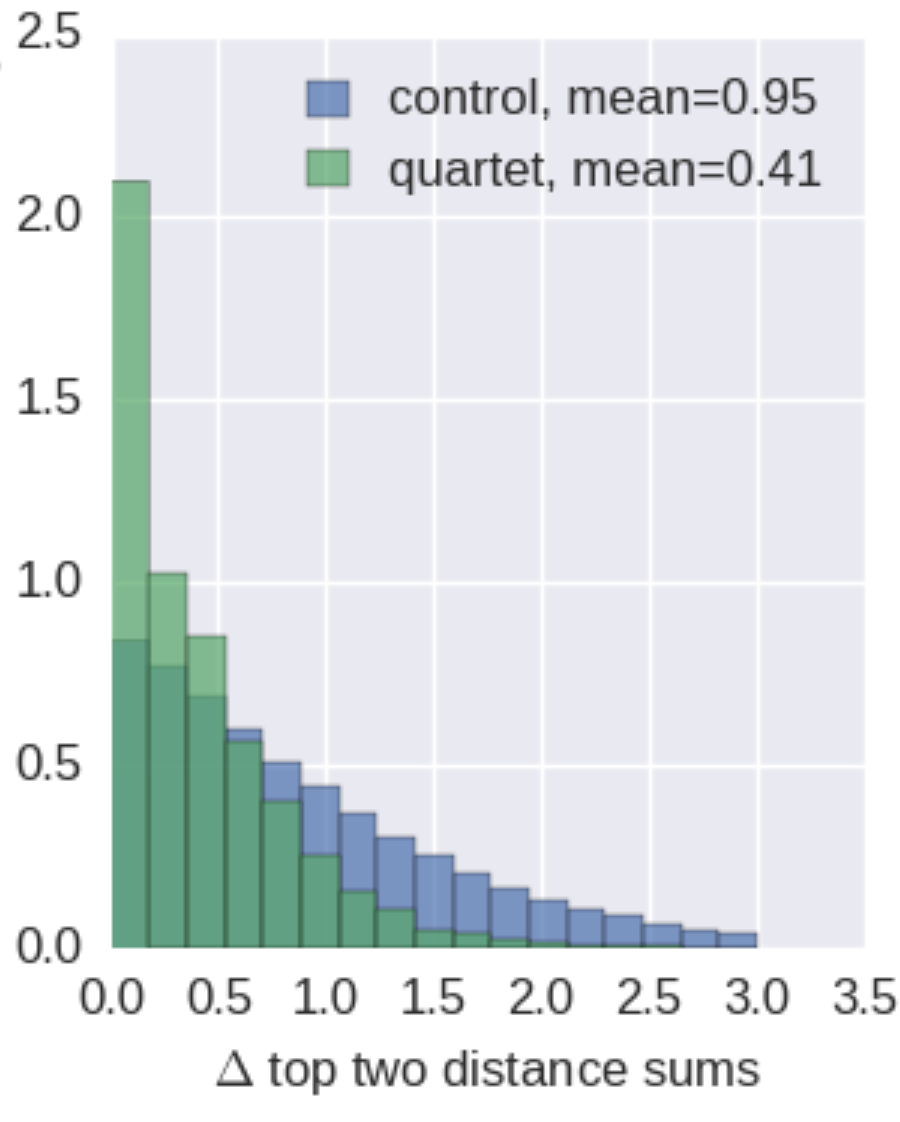
Rate of antigenic evolution
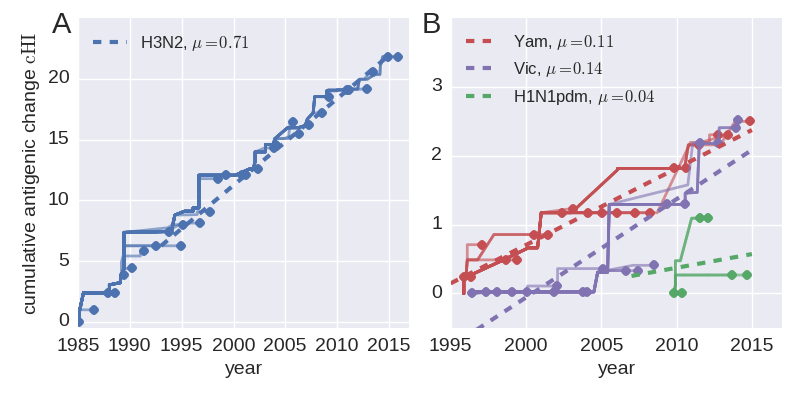
- Cumulative antigenic evolution since the root: $\sum_i d_i$
- A/H3N2 evolves faster antigenically
- A/H3N2 has a more rapid population turn-over
- Proportion of children is high in B vs A/H3N2 infections
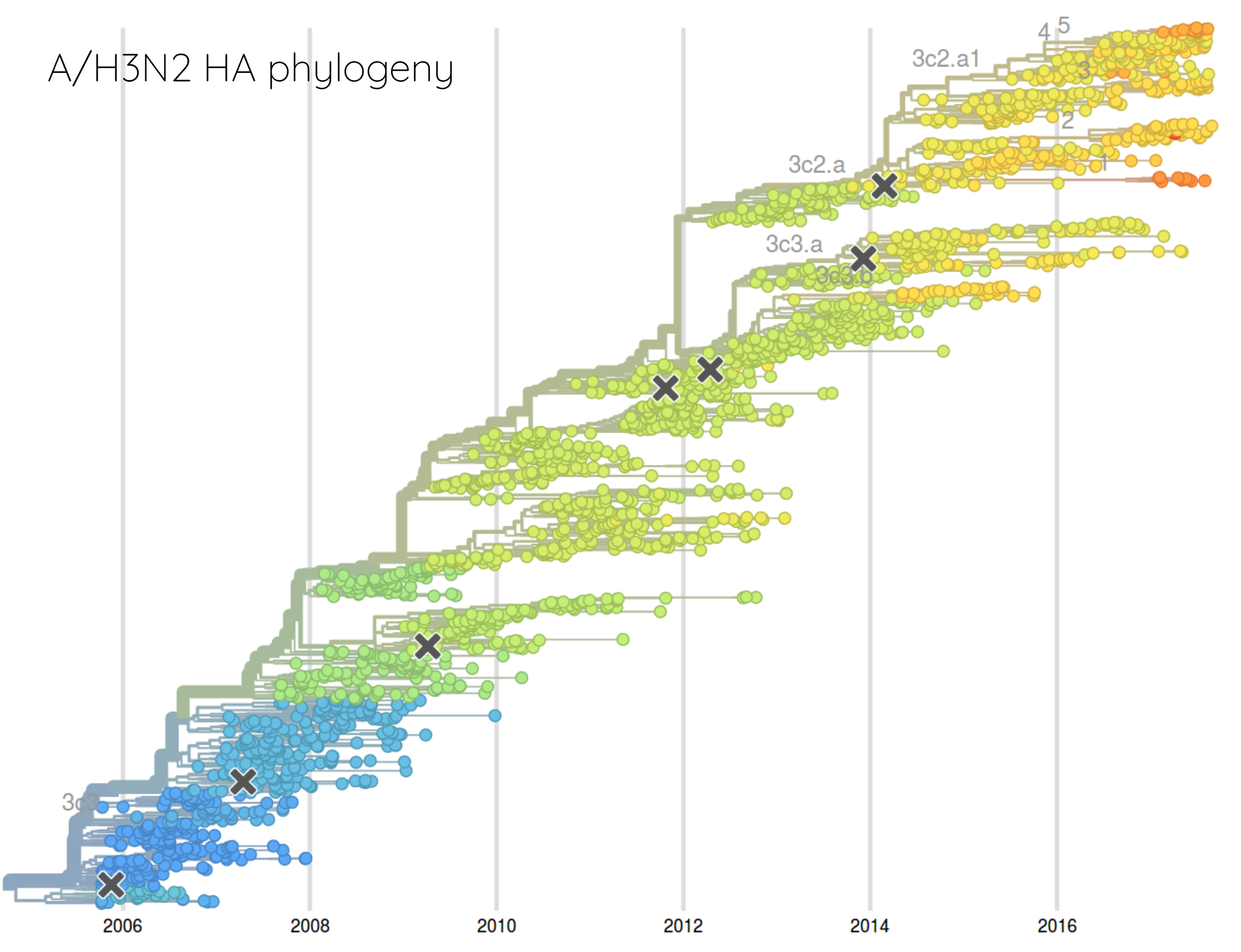
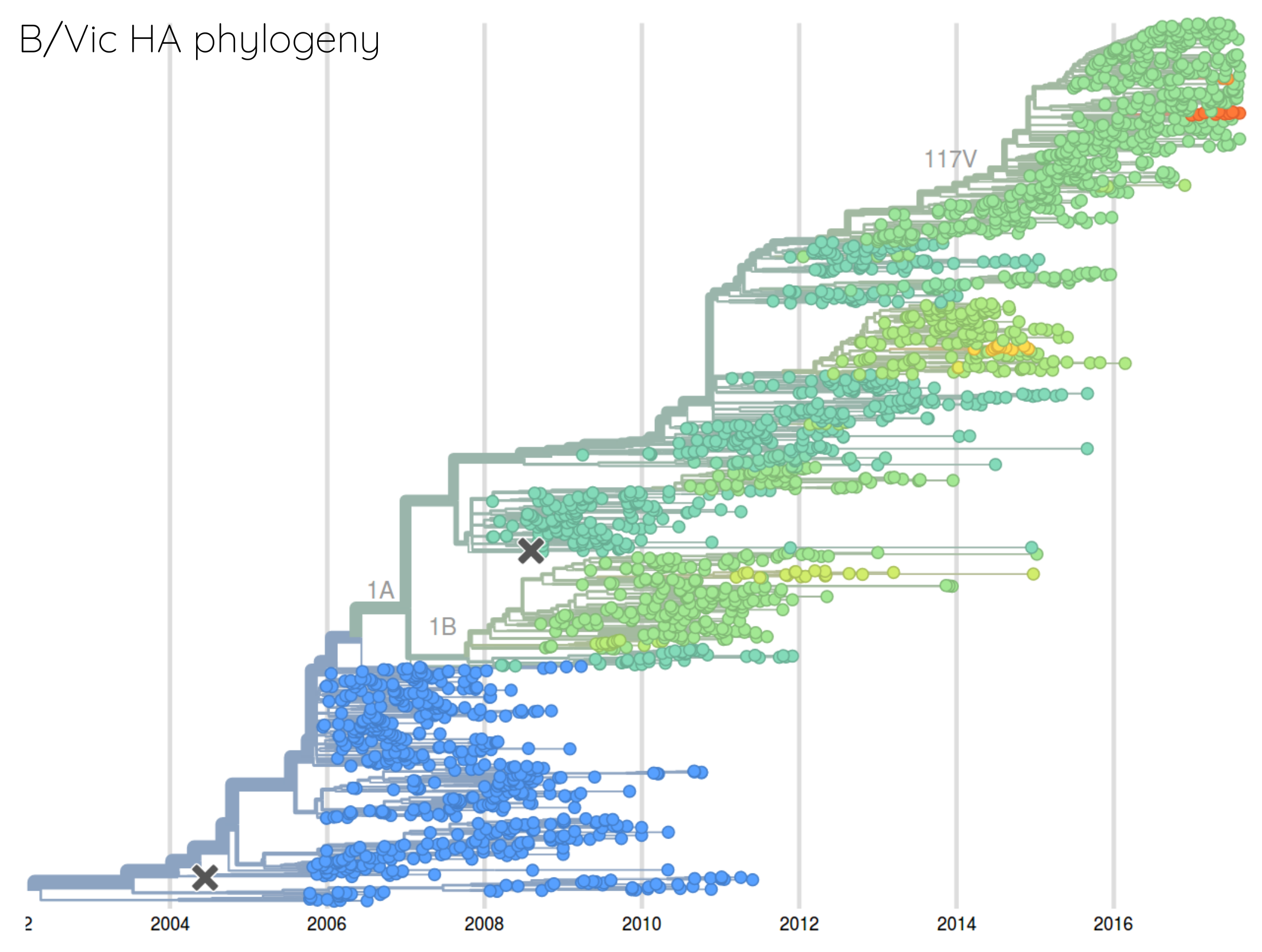
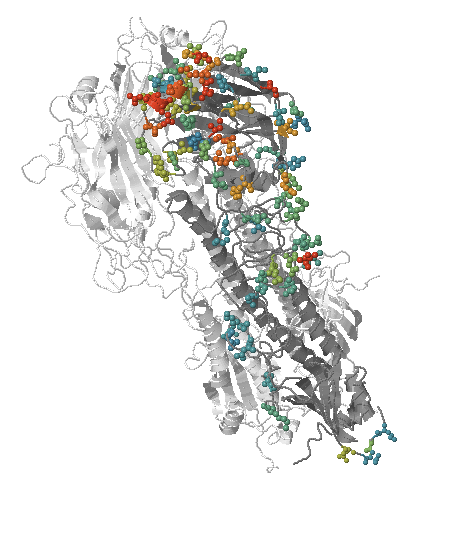
How many sites are involved?

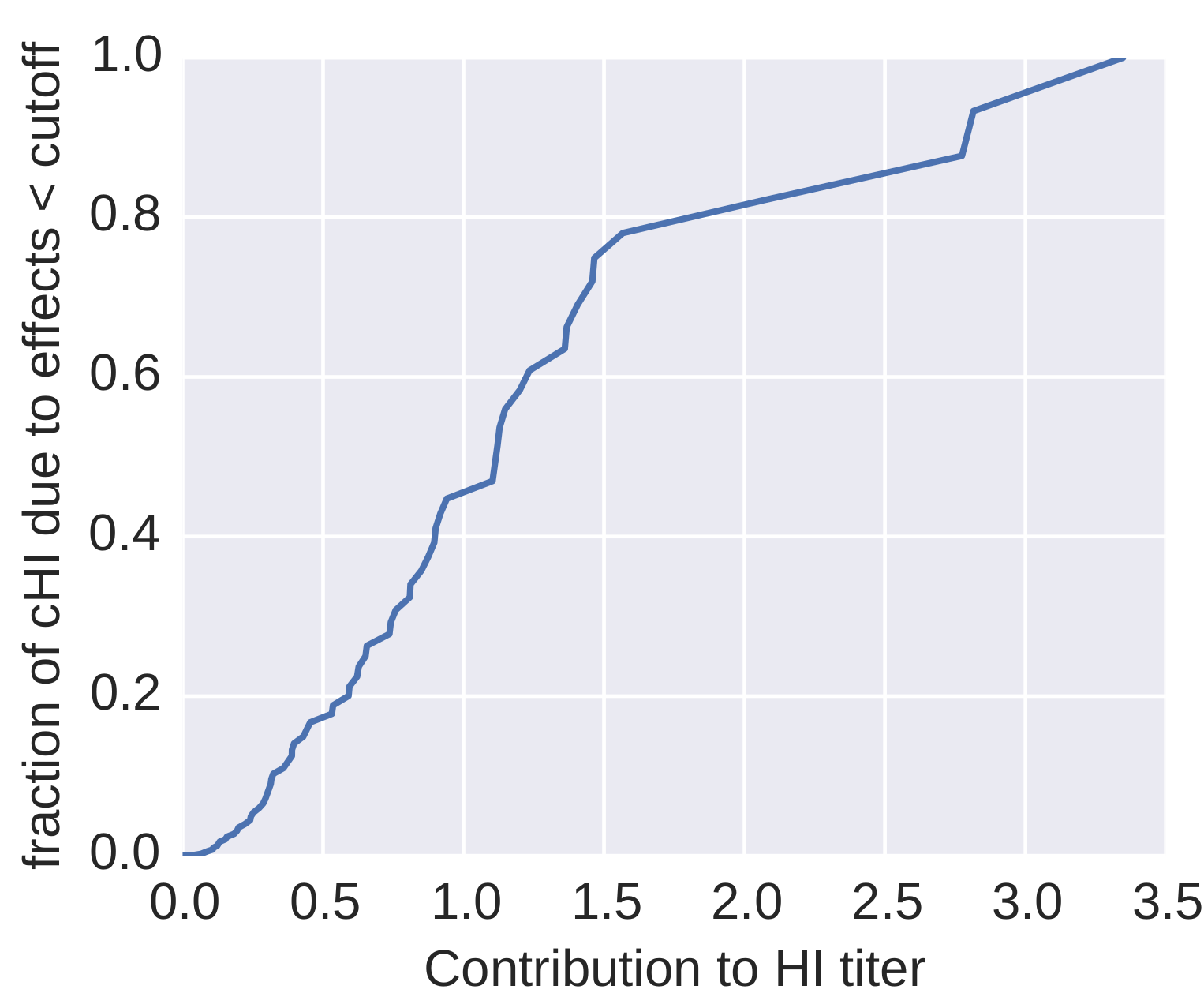
| Mutation | effect |
| K158N/N189K | 3.64 |
| K158R | 2.31 |
| K189N | 2.18 |
| S157L | 1.29 |
| V186G | 1.25 |
| S193F | 1.2 |
| K140I | 1.1 |
| F159Y | 1.08 |
| K144D | 1.08 |
| K145N | 0.91 |
| S159Y | 0.89 |
| I25V | 0.88 |
| Q1L | 0.85 |
| K145S | 0.85 |
| K144N | 0.85 |
| N145S | 0.85 |
| N8D | 0.73 |
| T212S | 0.69 |
| N188D | 0.65 |
Predicting successful influenza clades
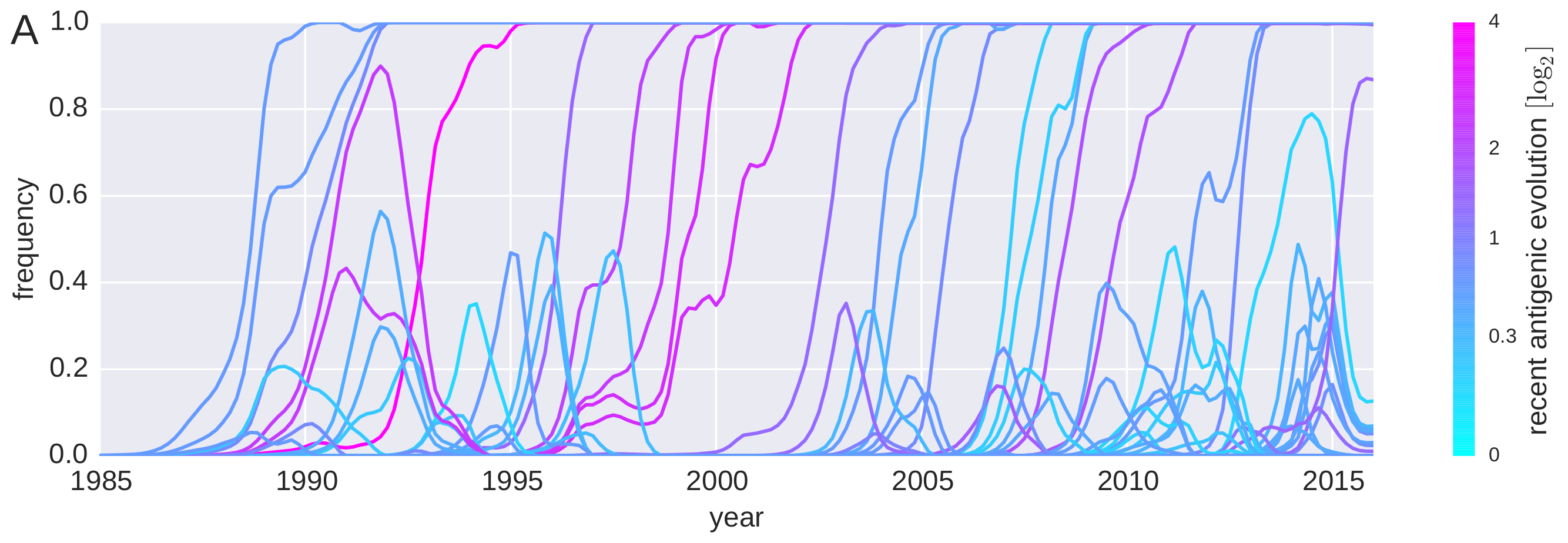
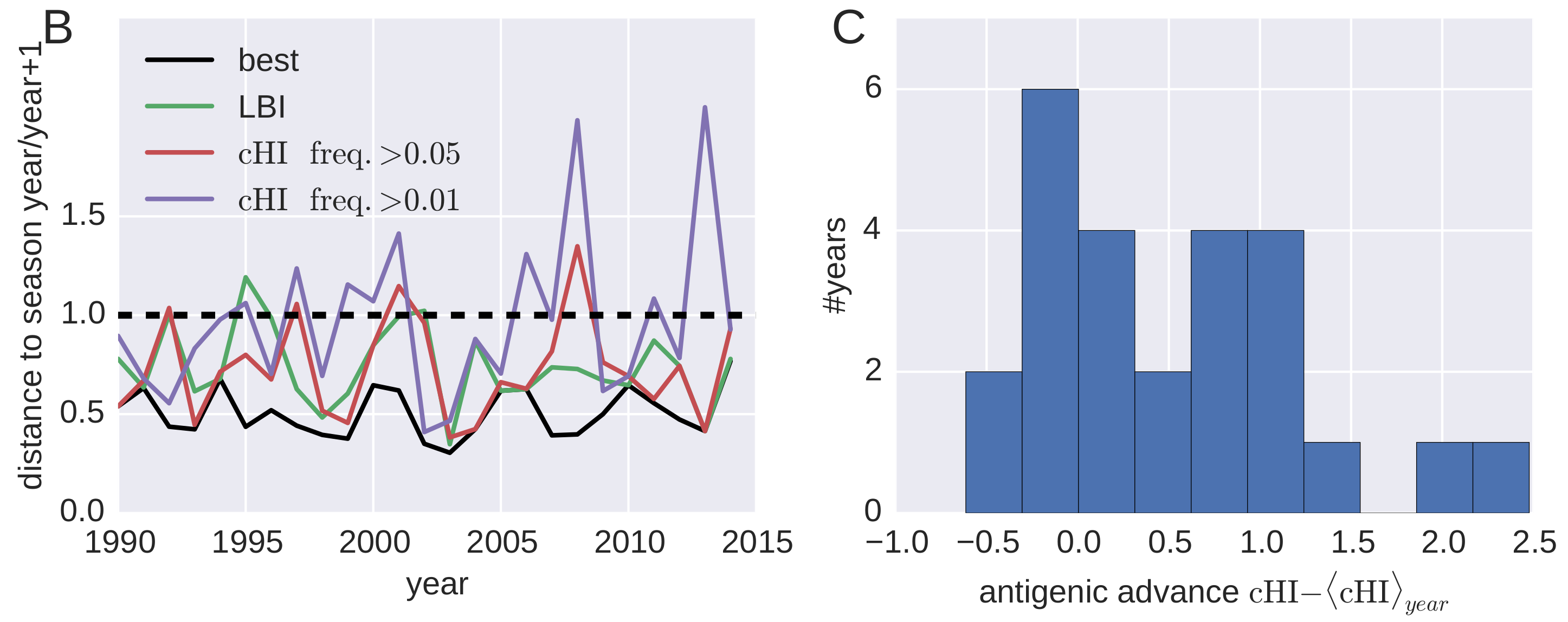
Predicting successful influenza clades
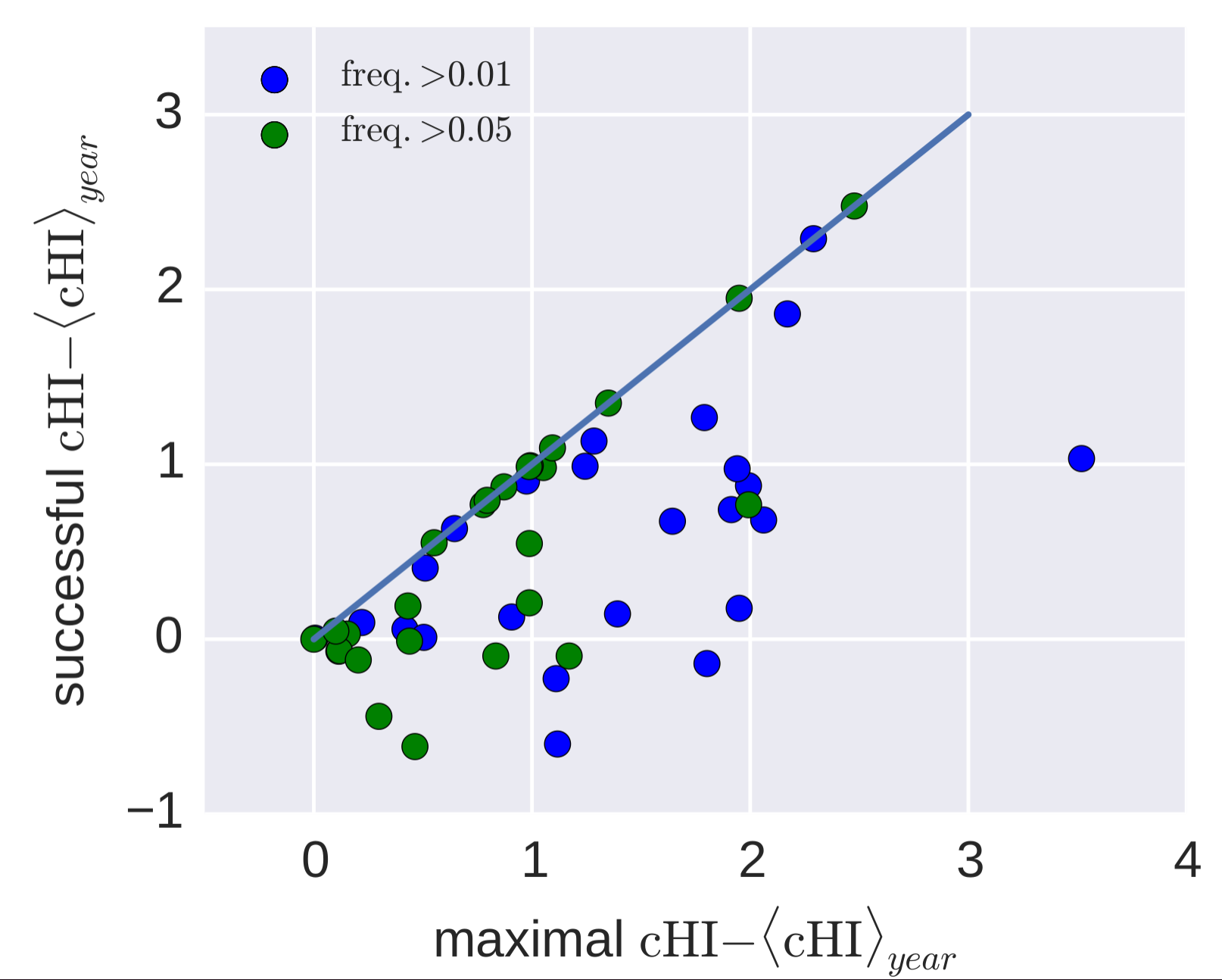
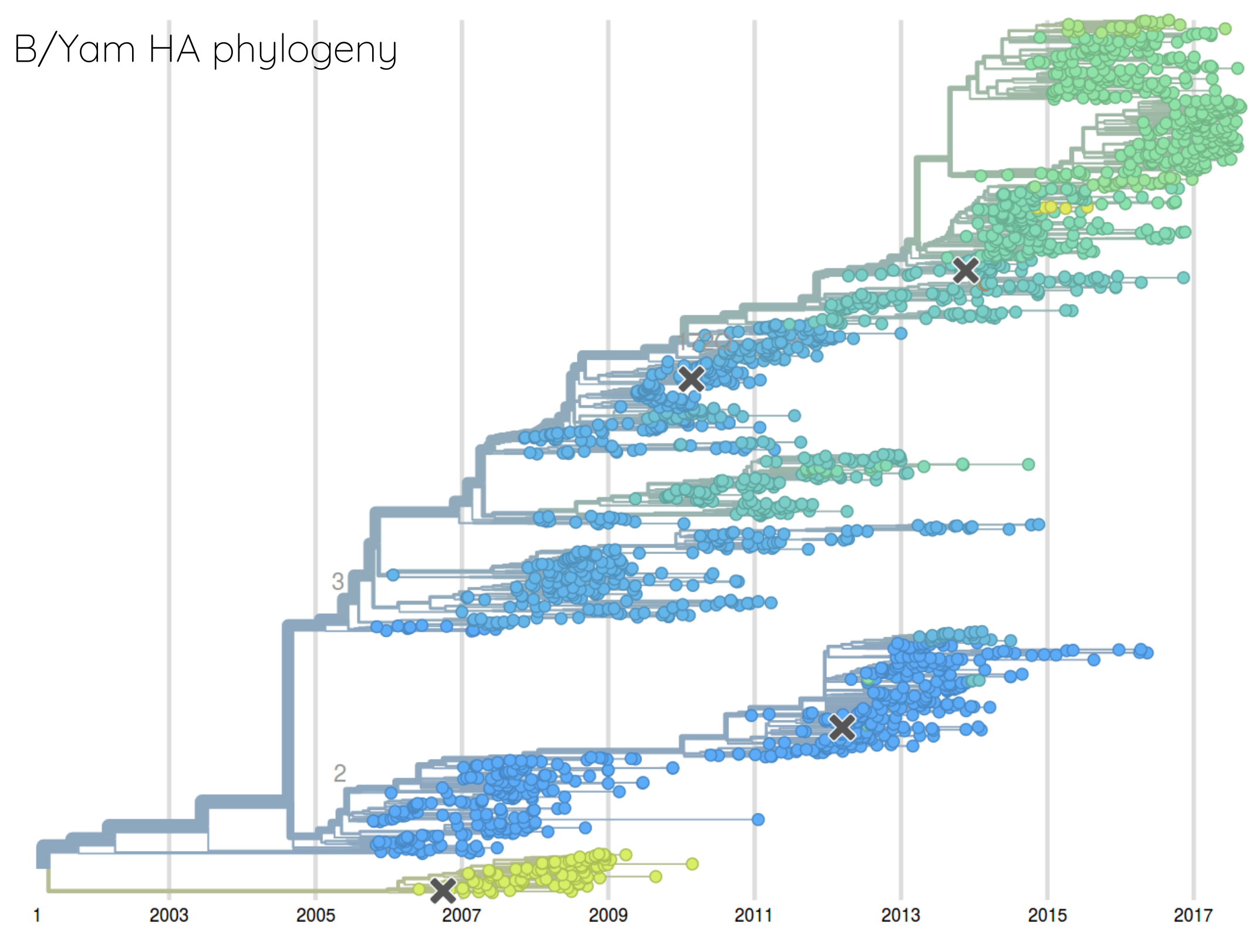
HI distances on the phylogenetic tree
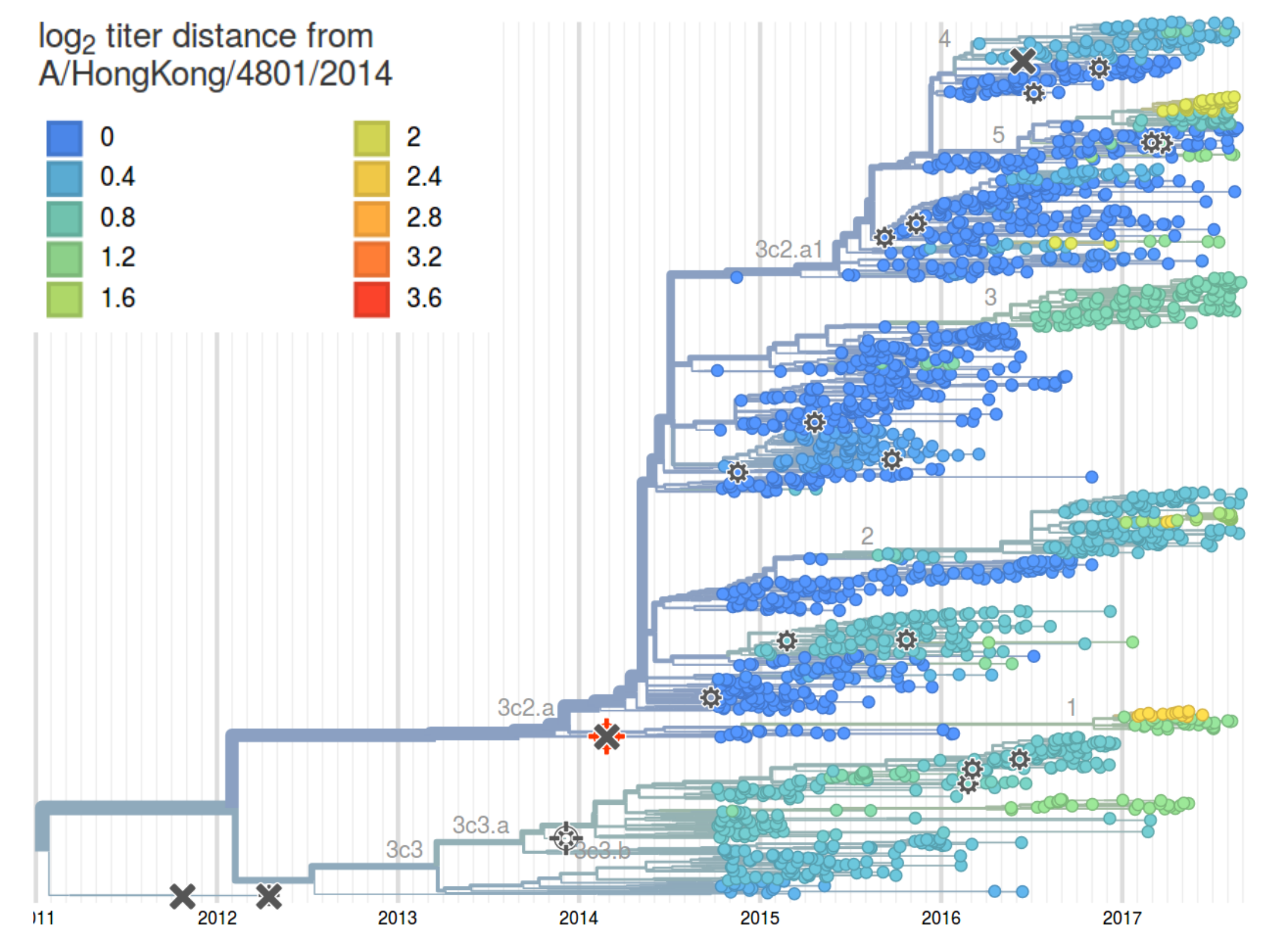
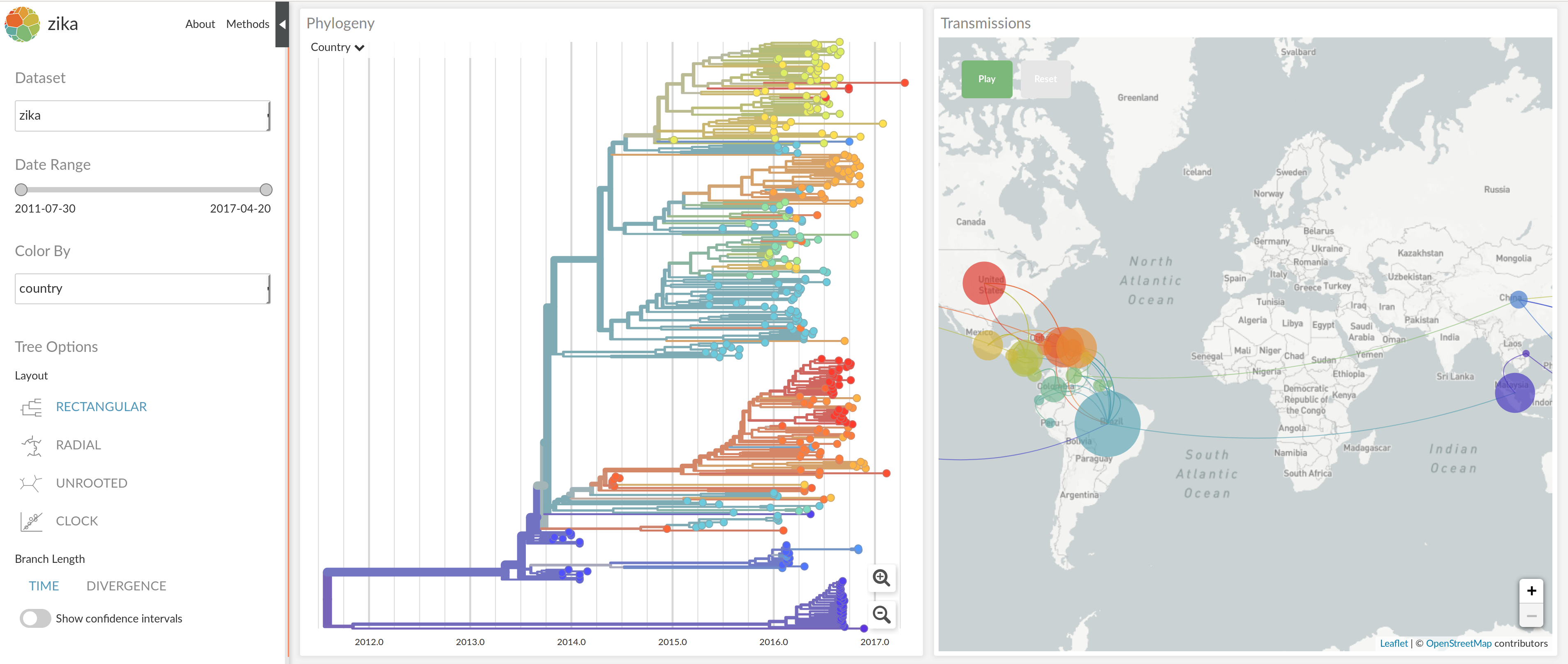
nextstrain.org
joint work with Trevor Bedford & his lab
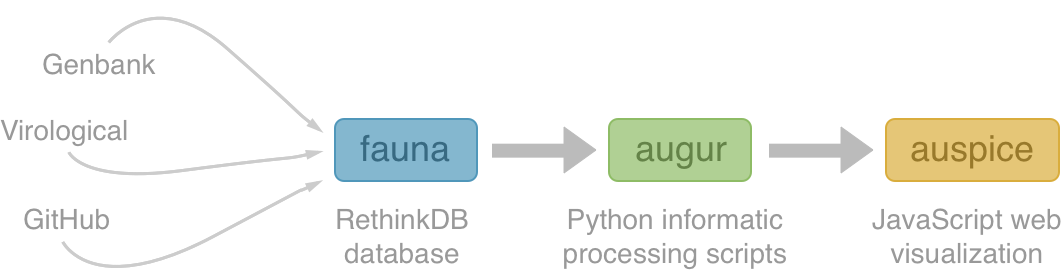
NextStrain architecture
Using treetime to rapidly compute timetrees
Summary
- Evolutionary biology can help track and fight disease
- Theory shows how to infer fit clades
- Future influenza population can be anticipated
- Automated real-time analysis can make up-to-date analysis available to every body
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
nextstrain.org





- Trevor Bedford
- Colin Megill
- Pavel Sagulenko
- Sidney Bell
- James Hadfield
- Wei Ding
