Within-host evolution of HIV and population genetics of rapid adaptation
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201802_IST.html
Evolution of HIV
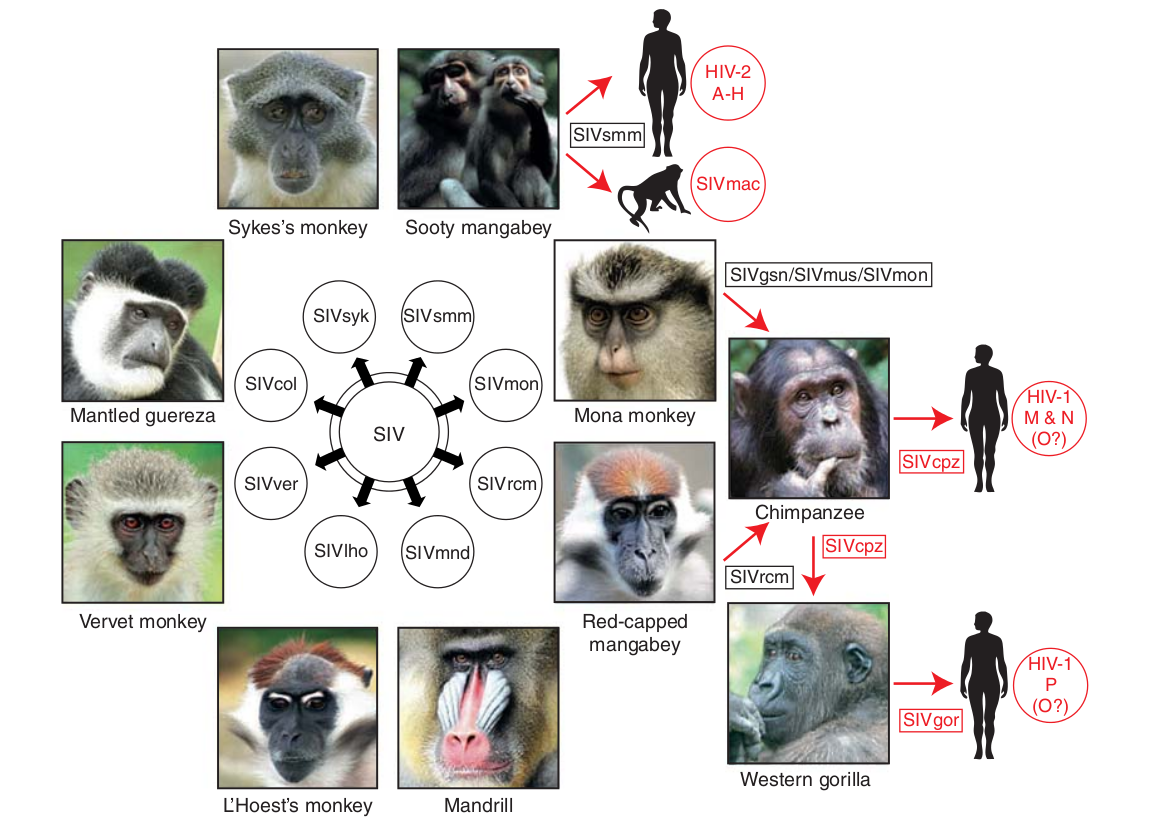
- Chimp → human transmission around 1900 gave rise to HIV-1 group M
- ~100 million infected people since
- subtypes differ at 10-20% of their genome
- HIV-1 evolves ~0.1% per year
HIV infection
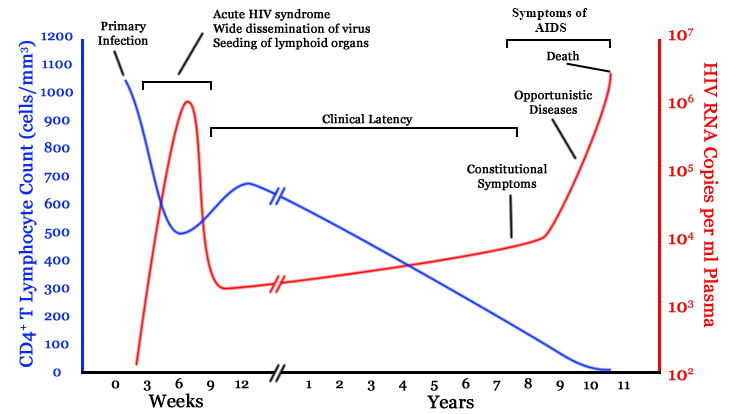

- $10^8$ cells are infected every day
- the virus repeatedly escapes immune recognition
- integrates into T-cells as
latent provirus
{kind=link}
HIV-1 evolution within one individual

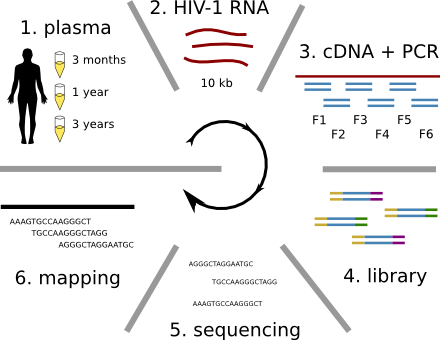

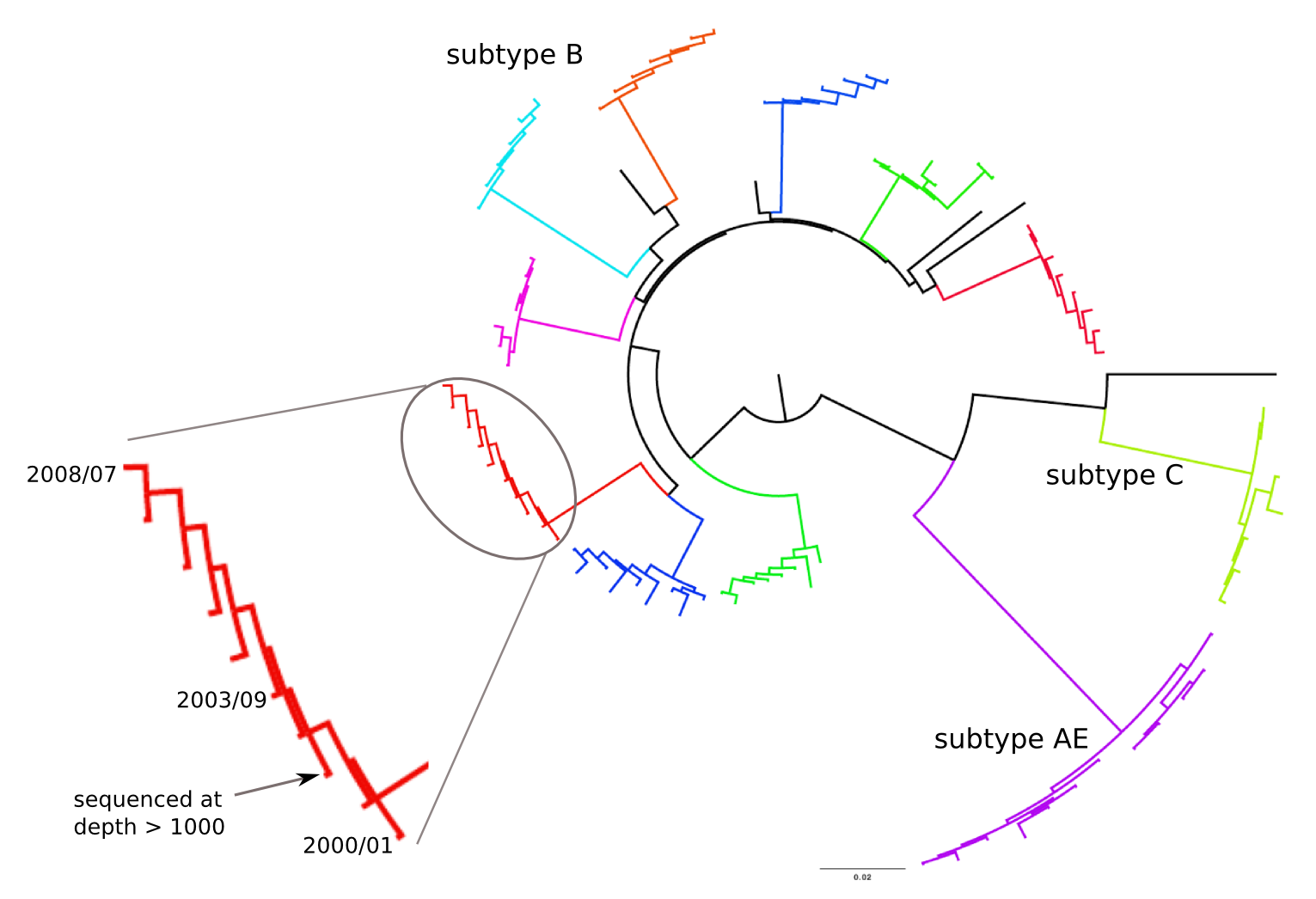
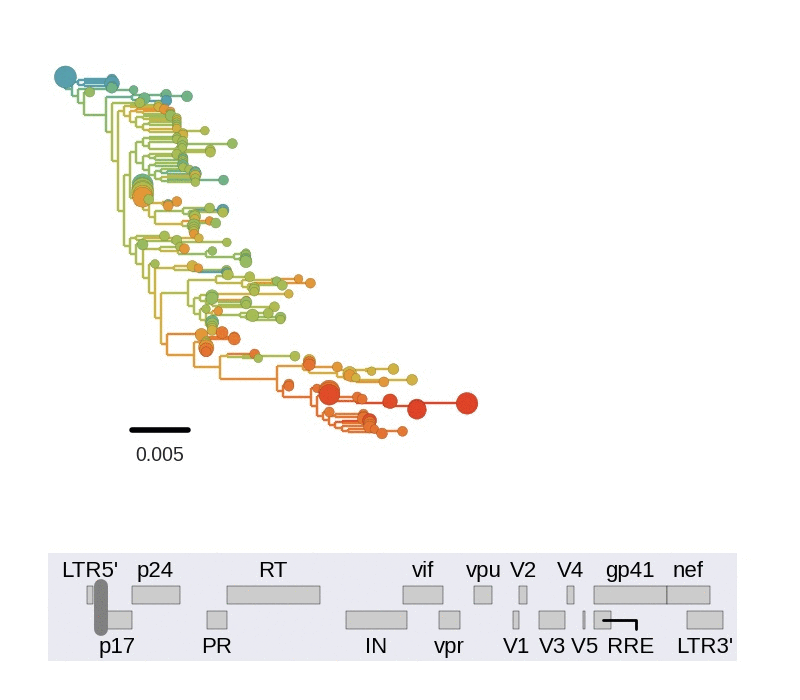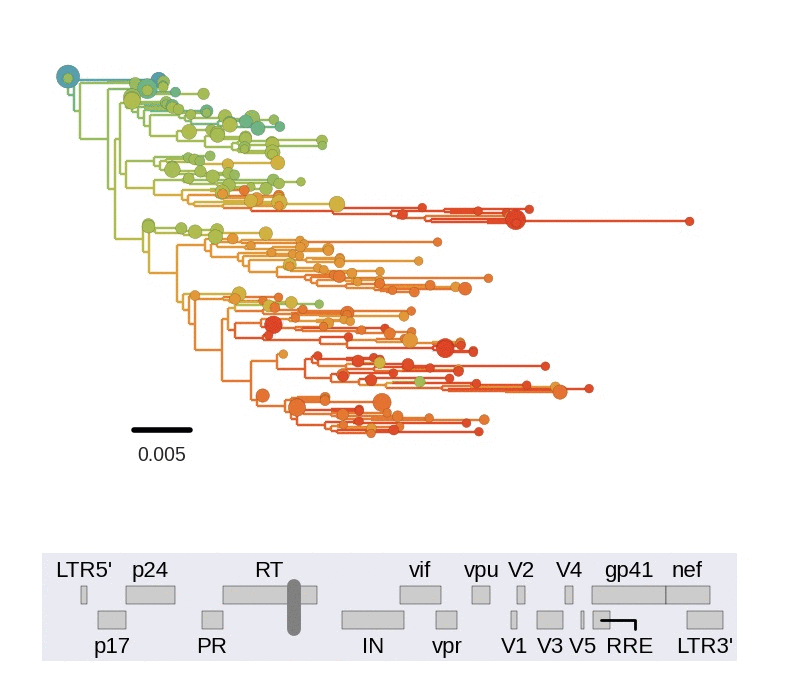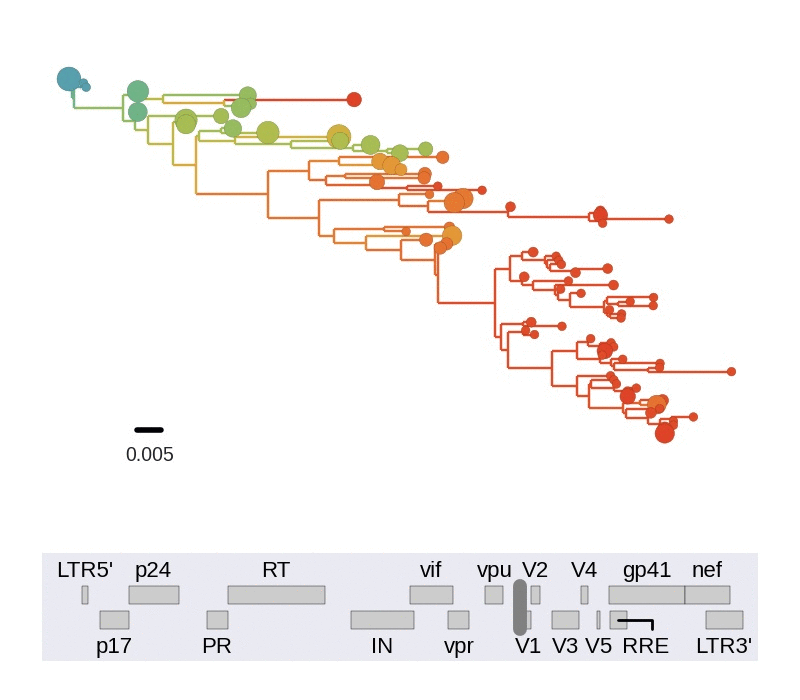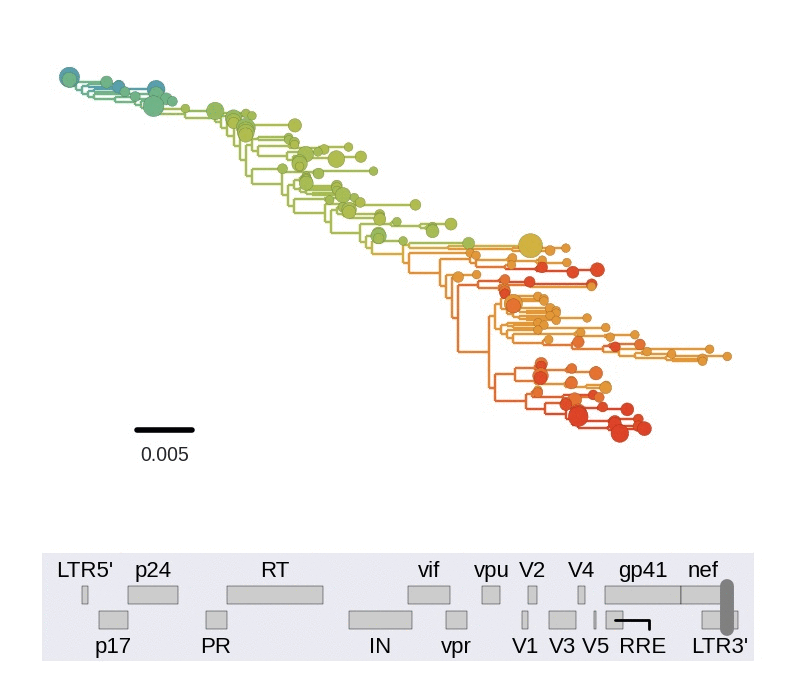
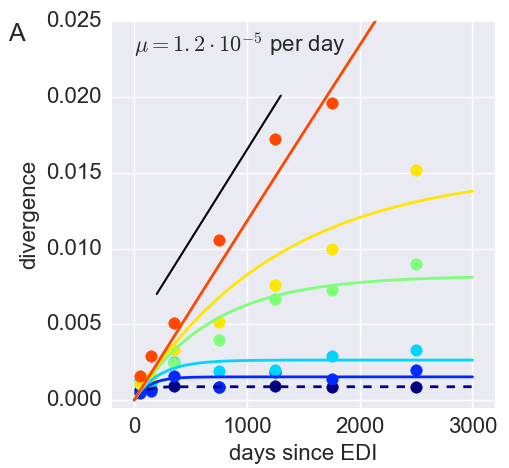
Mutation rates and diversity at neutral sites
 Zanini et al, Virus Evolution, 2017
Zanini et al, Virus Evolution, 2017
Frequent reversion of previously beneficial mutations
- HIV escapes immune systems
- most mutations are costly
- humans selects for different mutations
- compensation or reversion?
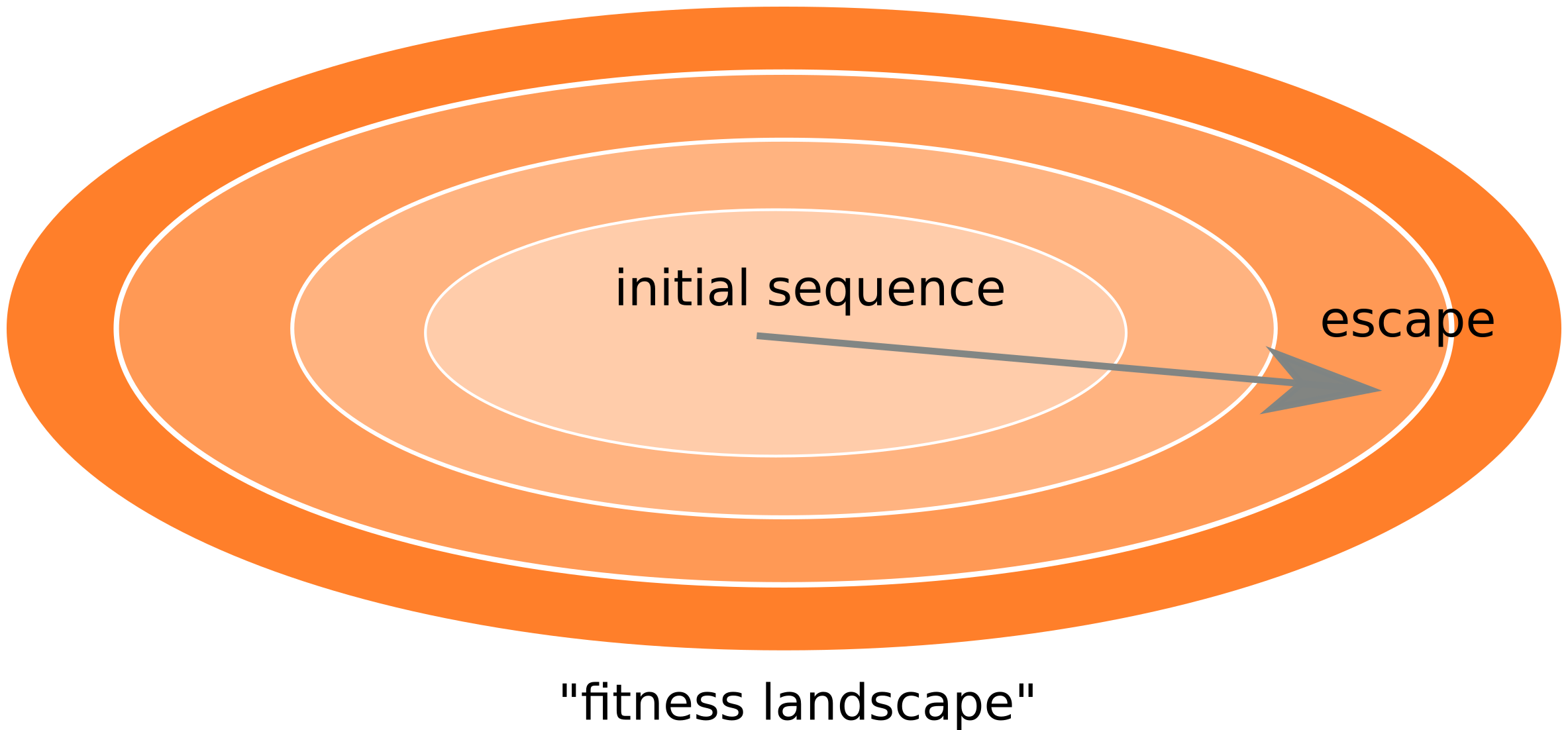
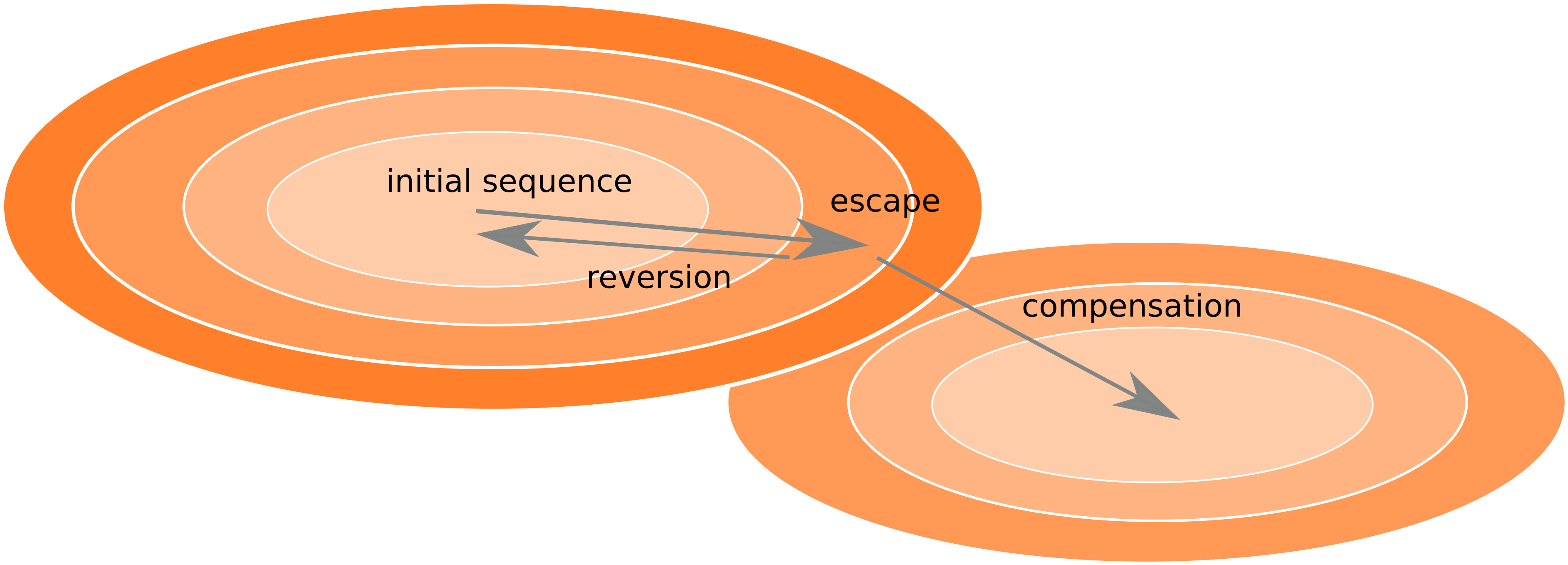
Inference of fitness costs
- mutation away from preferred state with rate $\mu$
- selection against non-preferred state with strength $s$
- variant frequency dynamics: $\frac{d x}{dt} = \mu -s x $
- equilibrium frequency: $\bar{x} = \mu/s $
- fitness cost: $s = \mu/\bar{x}$
Inference of fitness costs
- Frequencies of costly mutations decorrelate fast $\frac{d x}{dt} = \mu -s x $
- $\Rightarrow$ average many samples to obtain accurate estimates
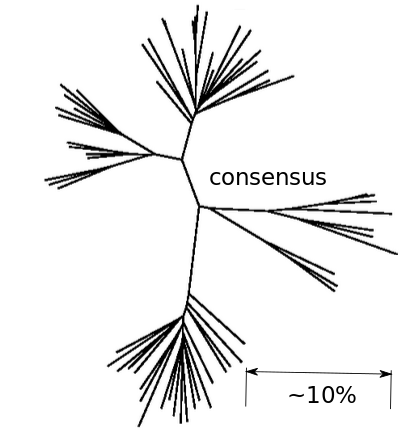
- Assumption: The global consensus is the preferred state
- Only use sites that initially agree with consensus
- Only use sites that don't chance majority nucleotide

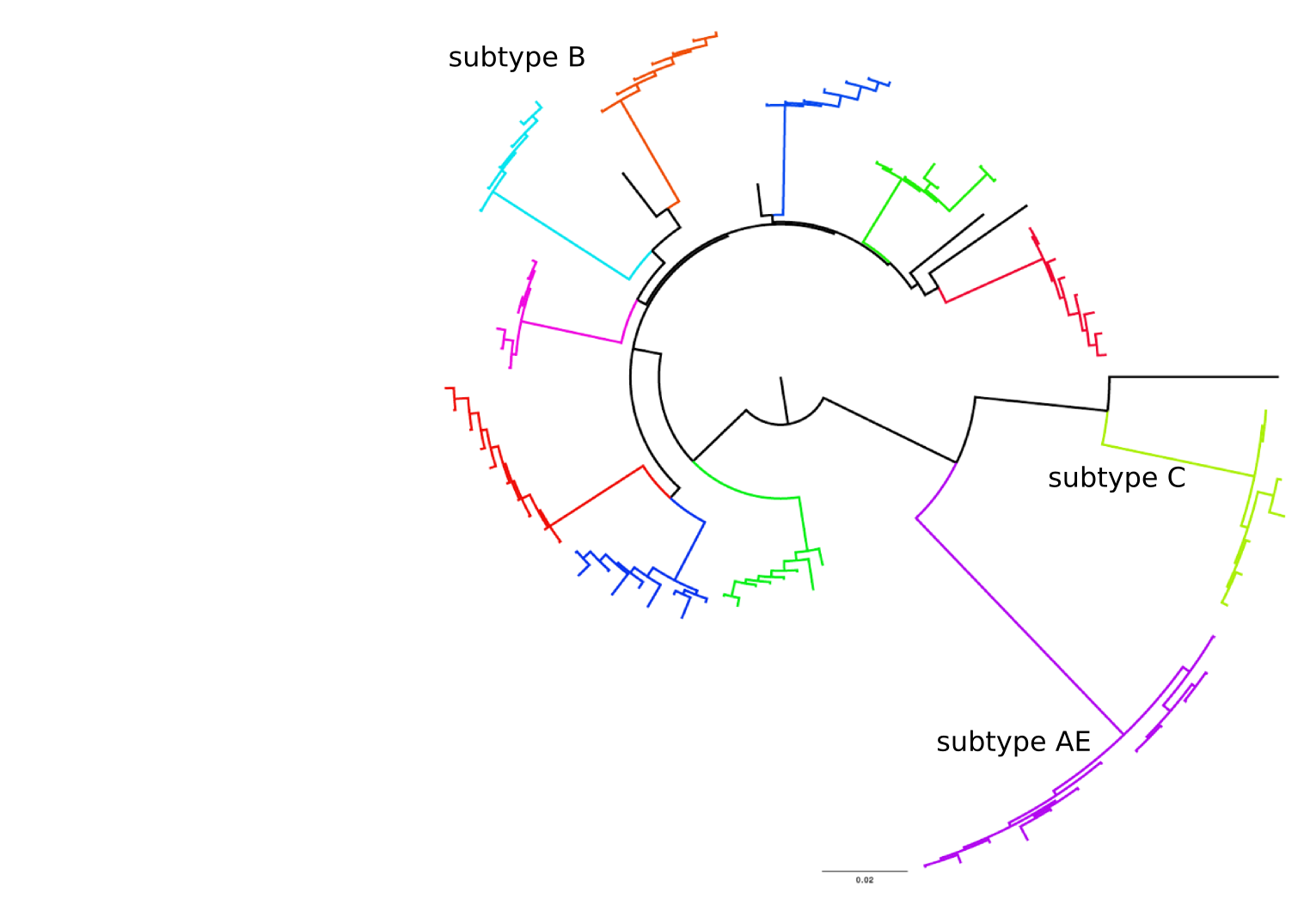
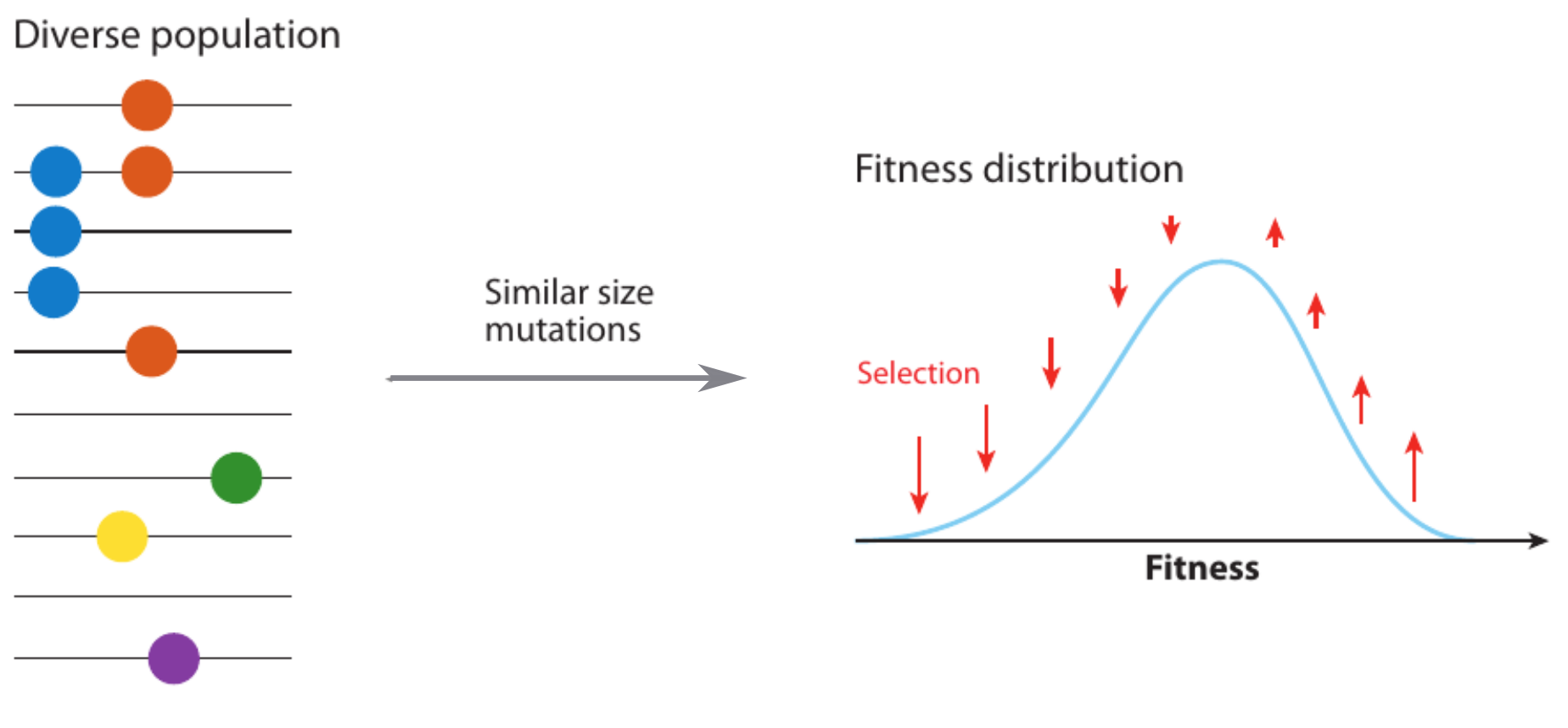
Fitness landscape of HIV-1
Zanini et al, Virus Evolution, 2017Selection on RNA structures and regulatory sites
Zanini et al, Virus Evolution, 2017The distribution of fitness costs
Zanini et al, Virus Evolution, 2017Fitness variation in rapidly adapting populations
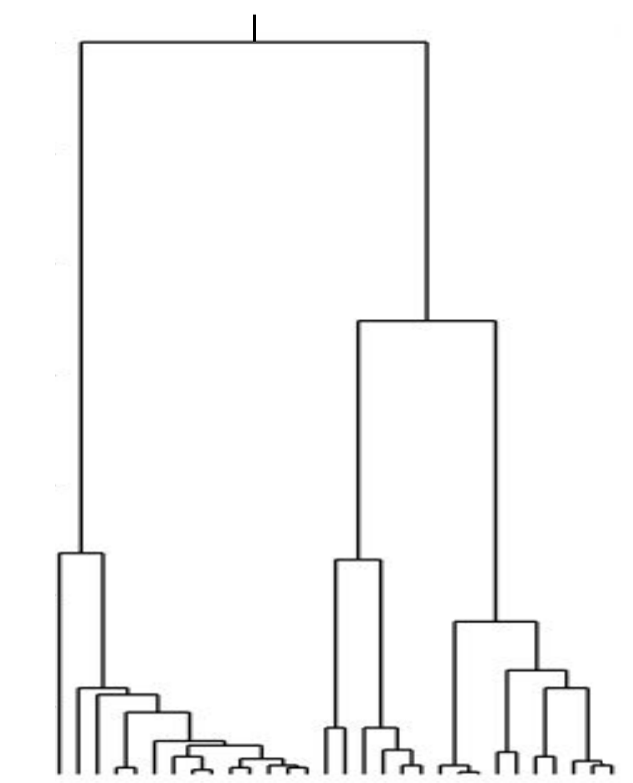
Neutral/Kingman coalescent
strong selection
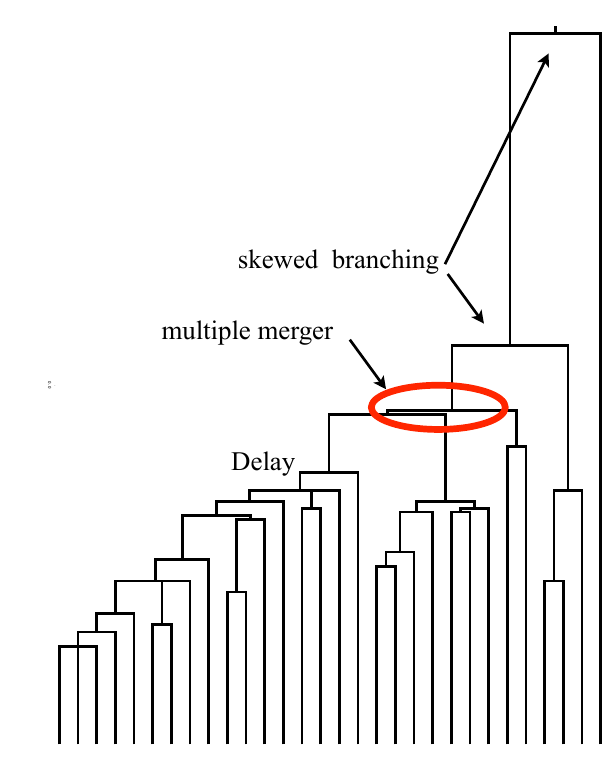
Bolthausen-Sznitman Coalescent
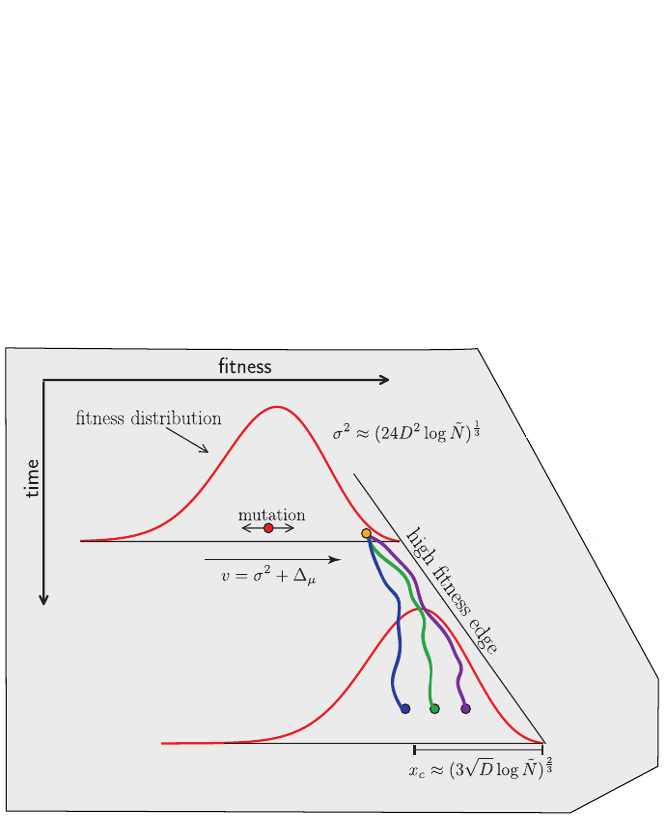
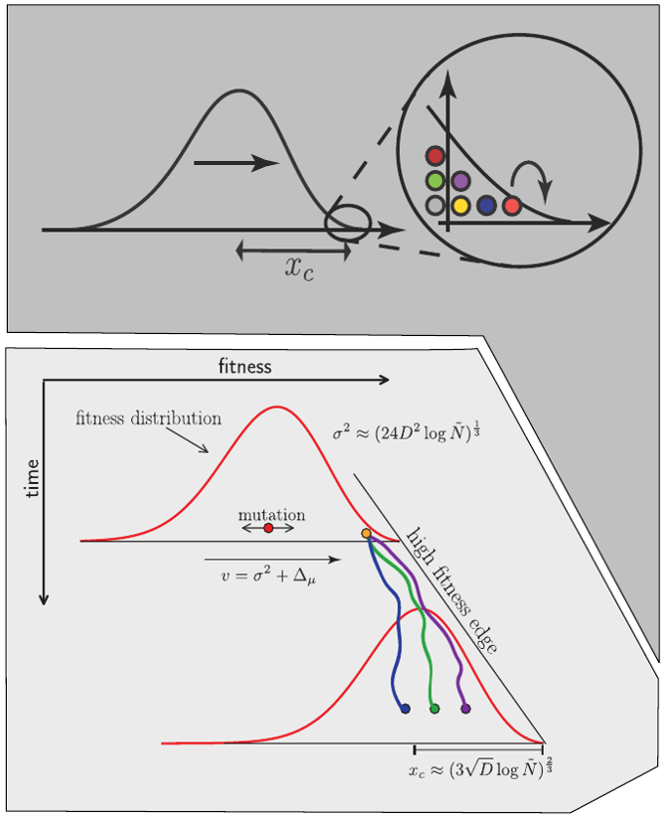
Traveling waves and the Bolthausen-Snitman coalescent
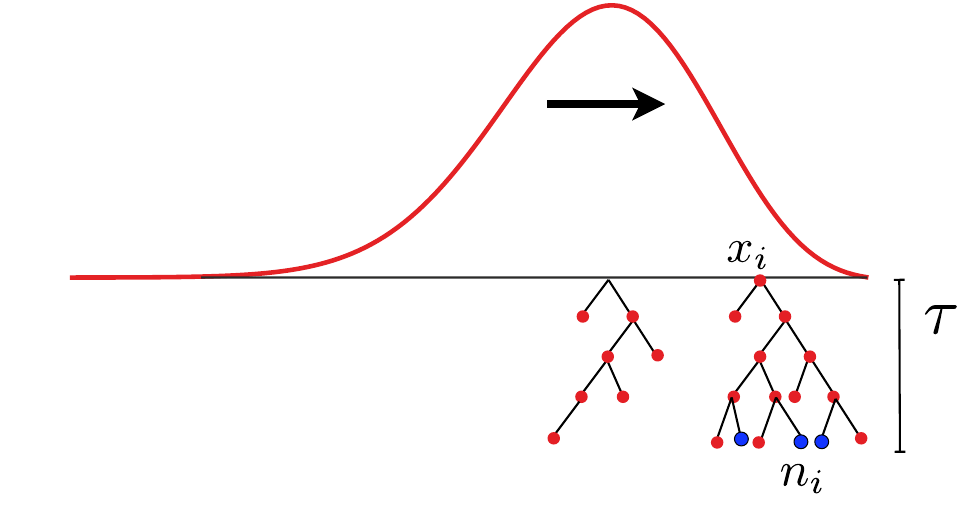
- Branching process approximation: $P(n_i, t|x_i)$
-
Does a sample (blue dots) have a common ancestor $\tau$ generations ago?
$\quad Q_b = \langle \sum_i \left(\frac{n_i}{\sum_j n_j}\right)^b\rangle \approx \frac{\tau-T_c}{T_c(b-1)} $ -
All other merger rates are also consistent with the Bolthausen-Sznitman coalescent:
$\quad\lambda_{b,k} = \frac{(k-2)!(b-k)!}{T_c (b-1)!}$
U-shaped polarized site frequency spectra
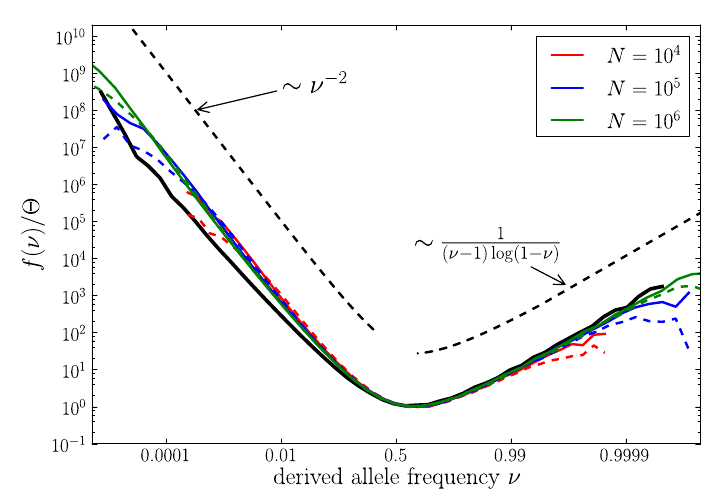

Universality -- adaptation and deleterious mutations
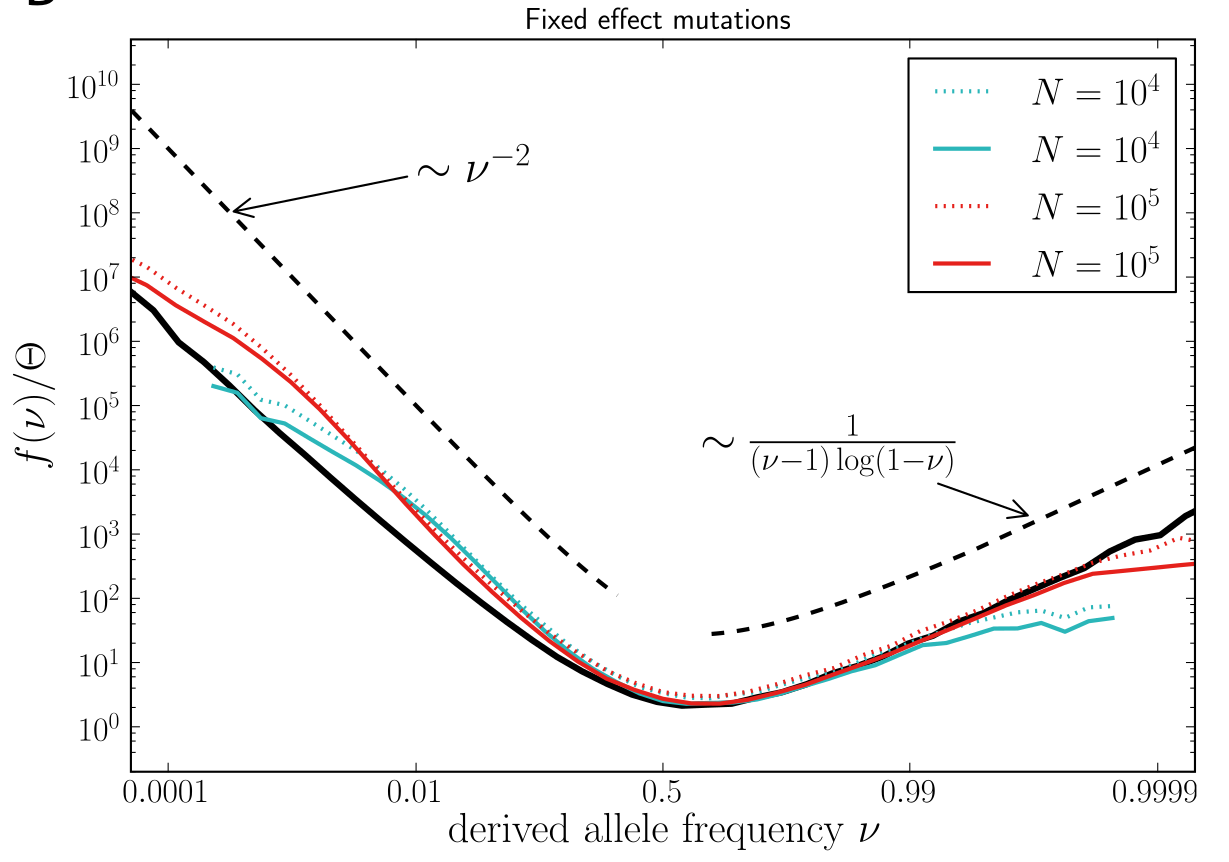
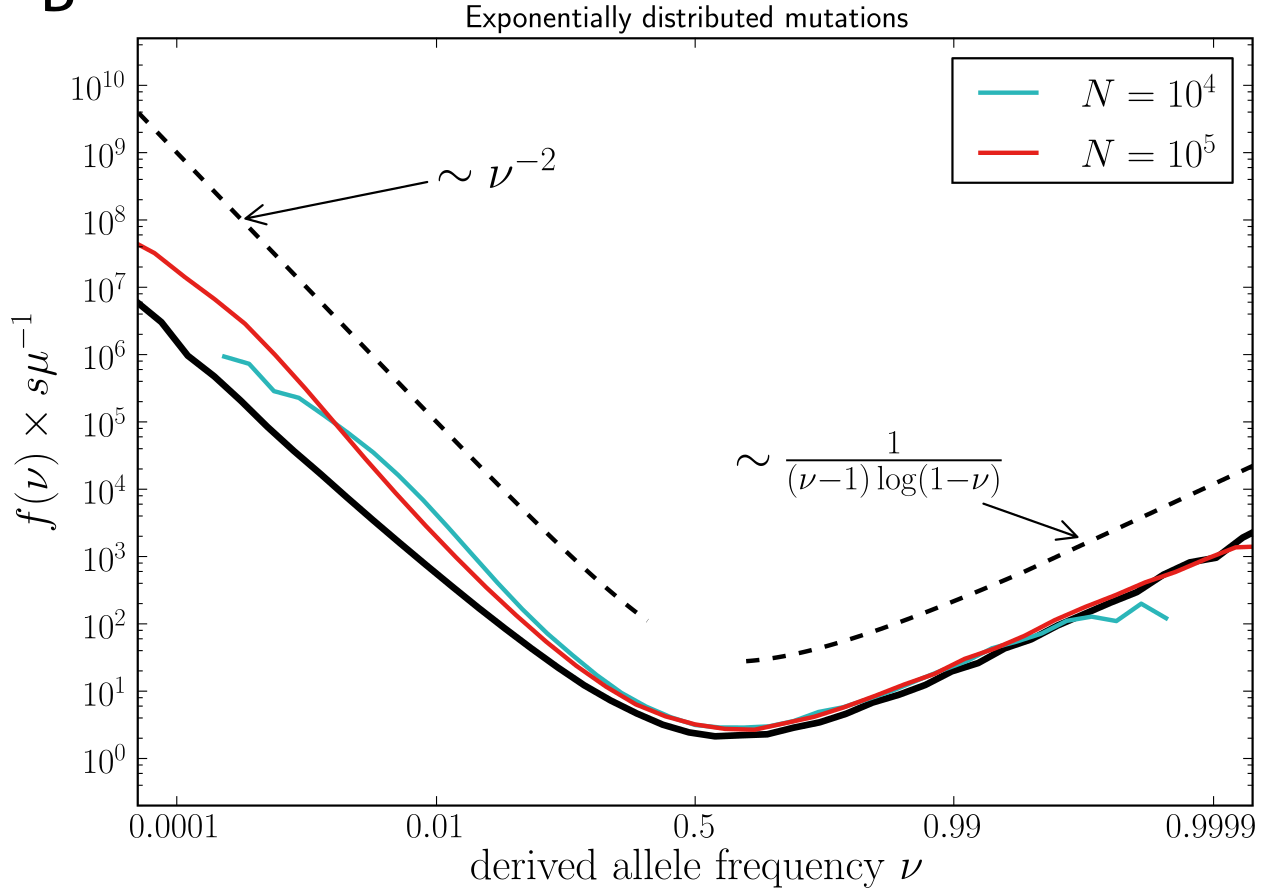
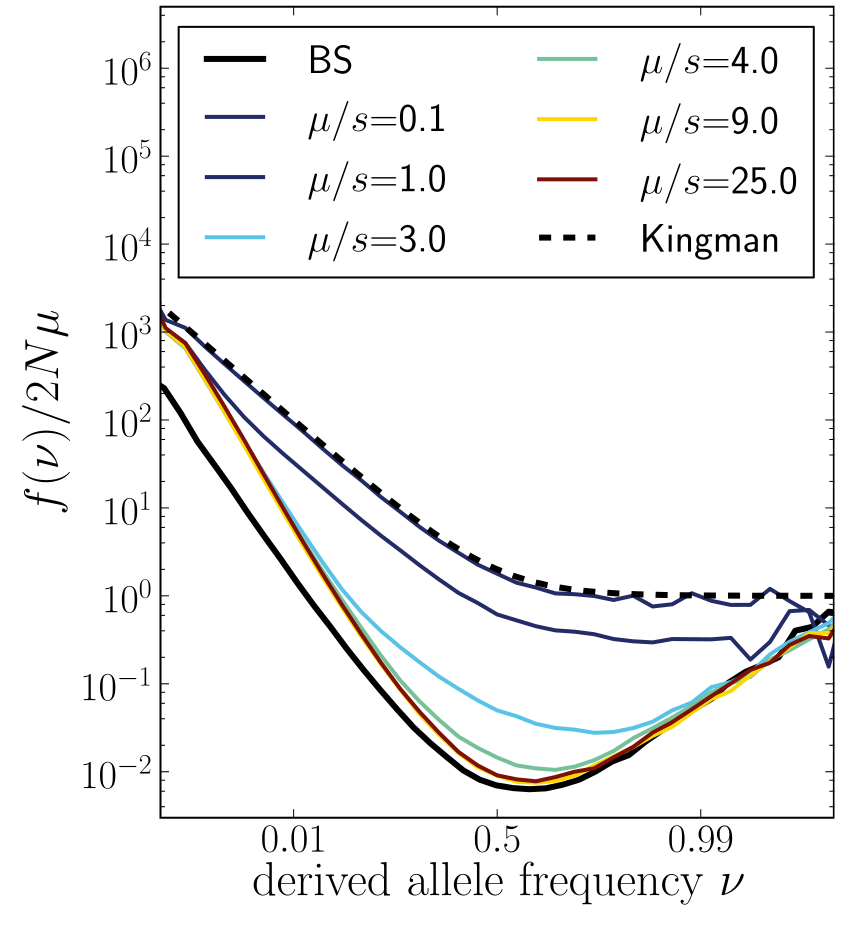
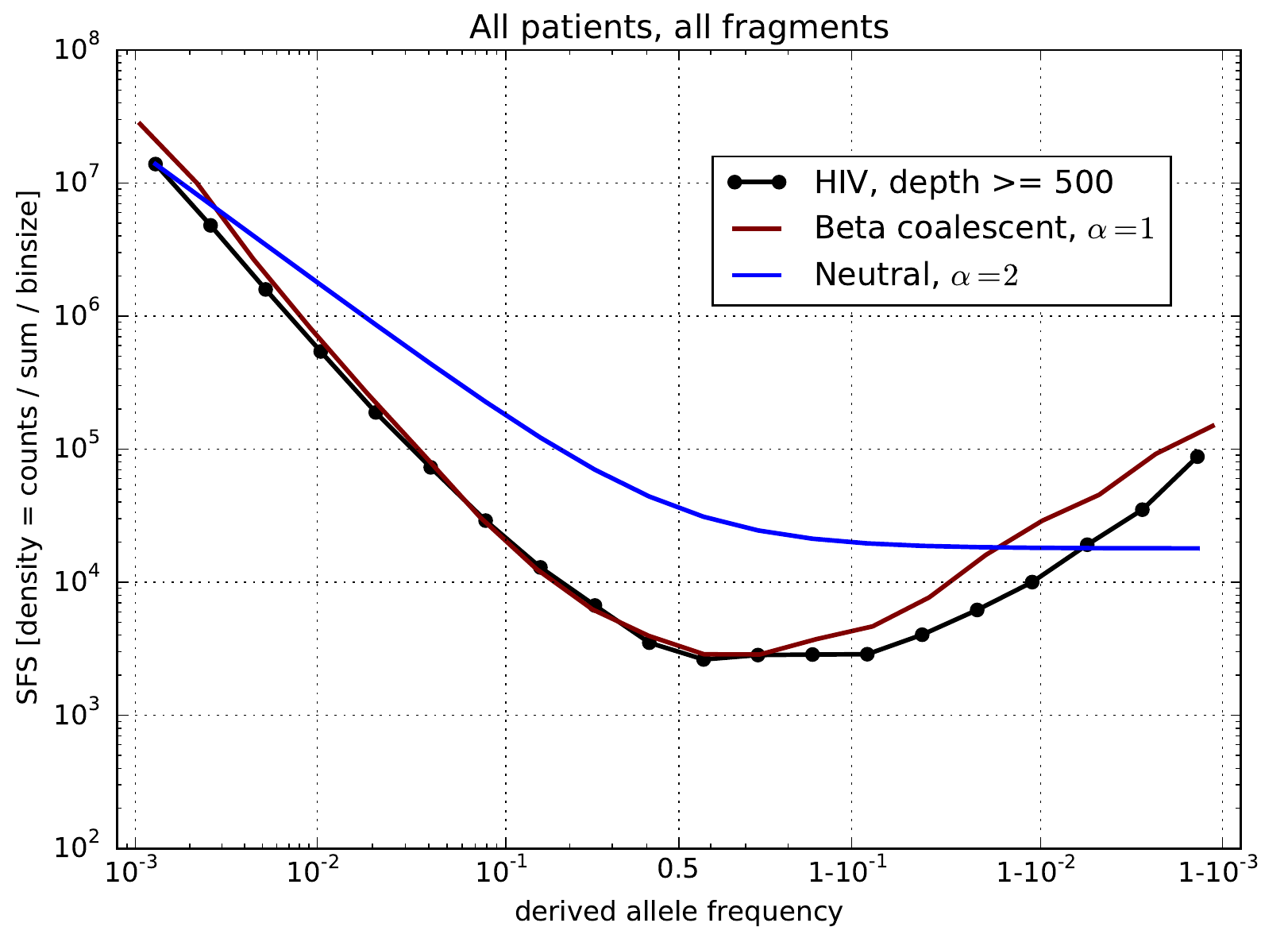 Zanini et al, eLife, 2016
Zanini et al, eLife, 2016
Extension to sexual populations
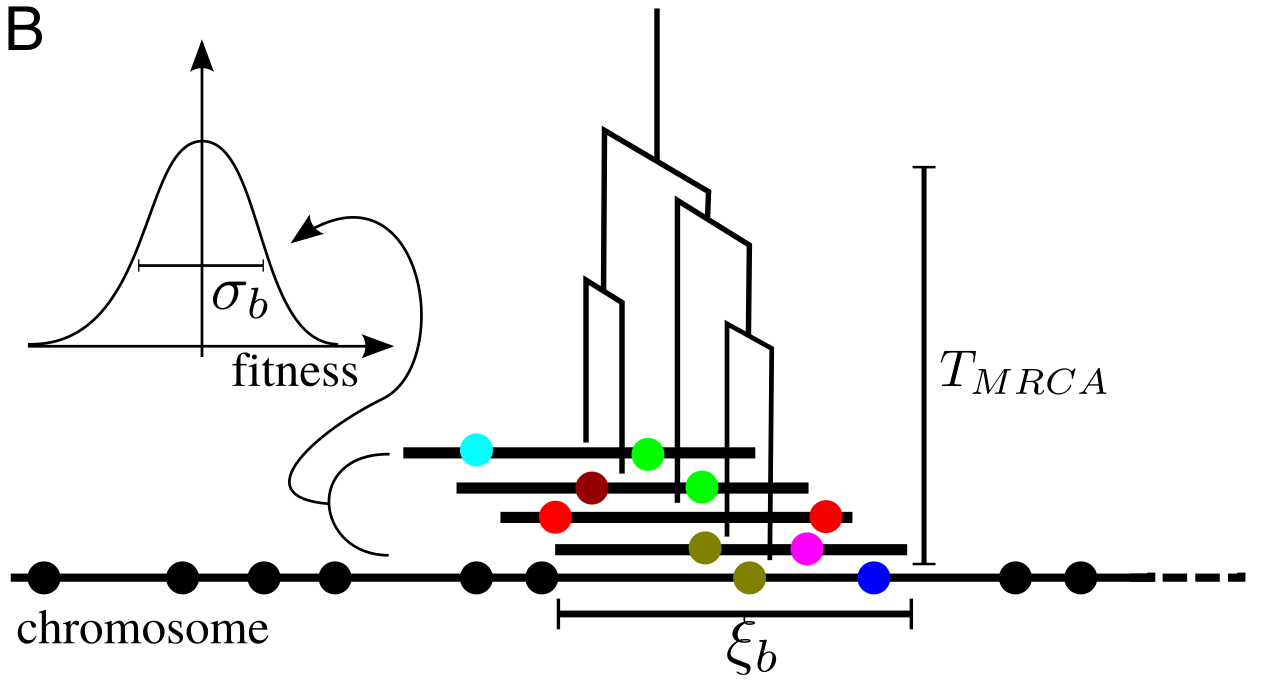
- $T_{MRCA}$ determined by $\sigma_b$
- Block length $\zeta_b$ is determined by $T_{MRCA}$
- Fitness variation $\sigma_b$ is determined by block length
$T_{MRCA}$ and SFS
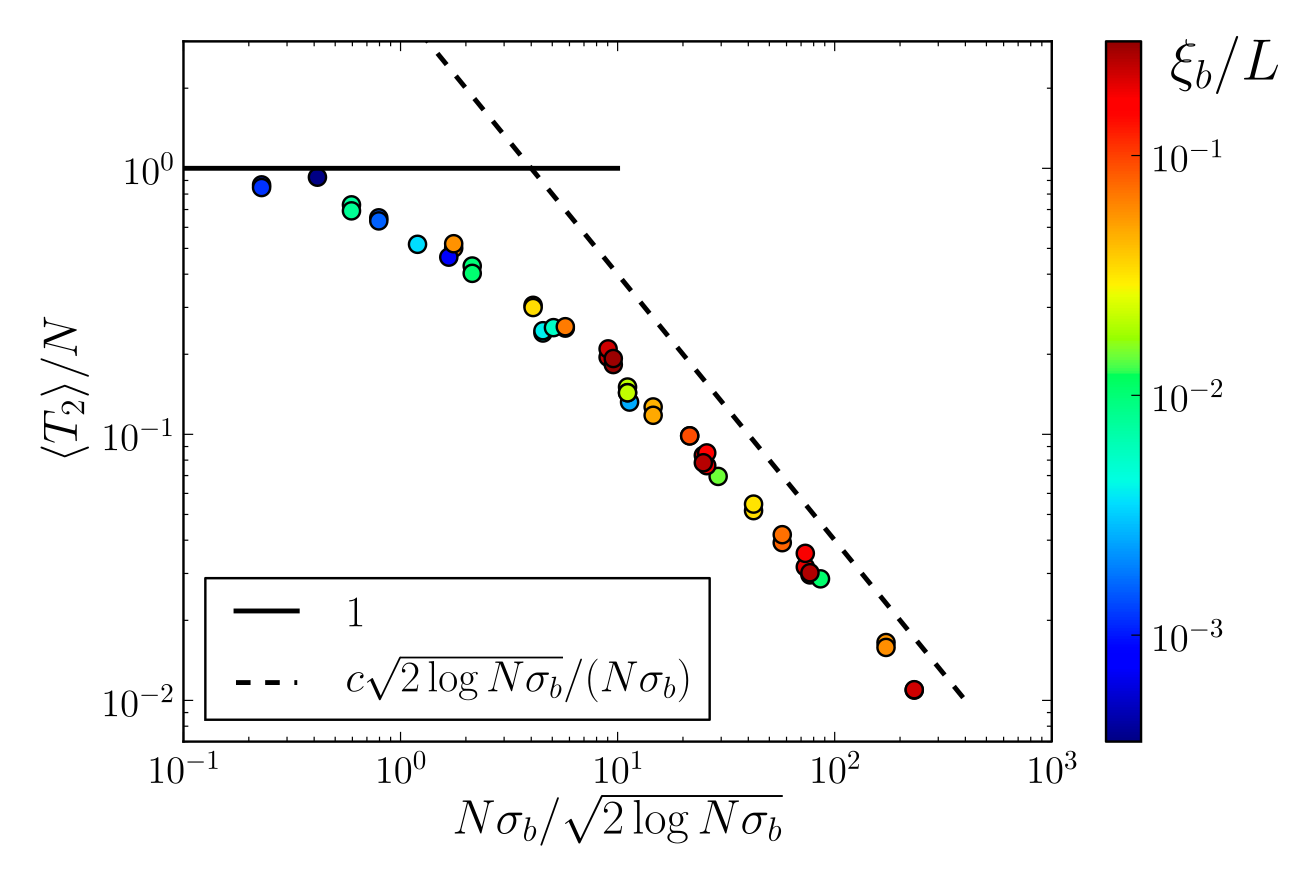
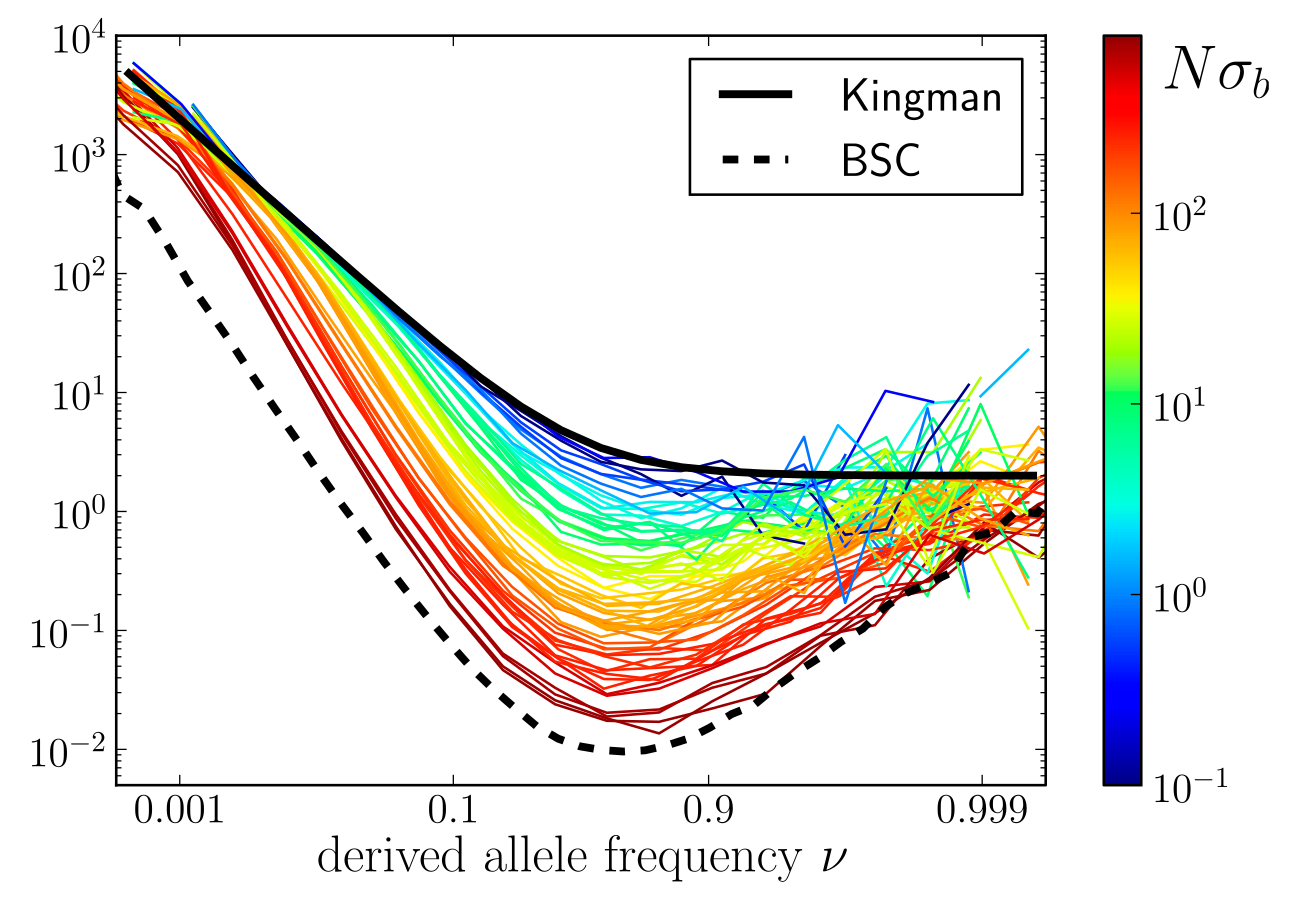
- Fitness diversity in block: $\sigma_b = \frac{\mu \langle s^2\rangle}{2\rho}$
- Qualitative change behavior around $N\sigma_b$
- Total rate of adaptation: $\sim L\sqrt{\rho \mu \langle s^2\rangle \log N}$
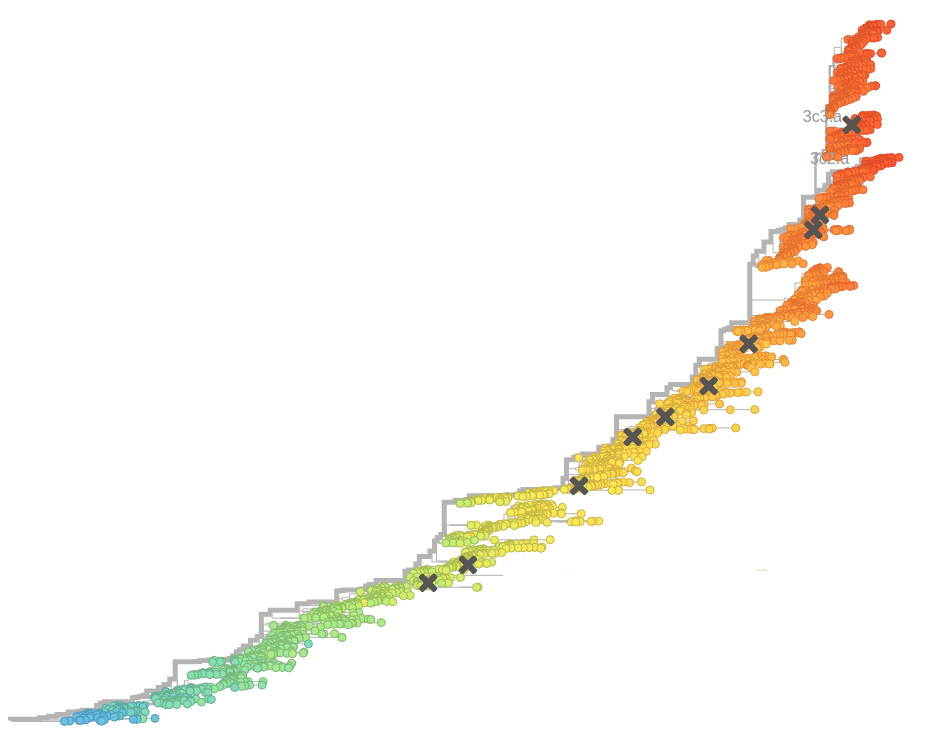
Bursts in a tree ↔ high fitness genotypes
Can we read fitness of a tree?
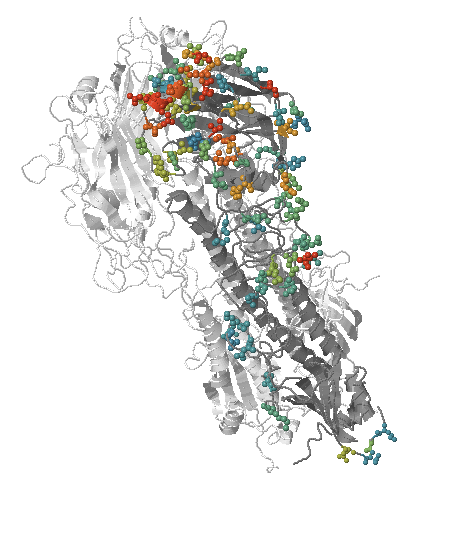
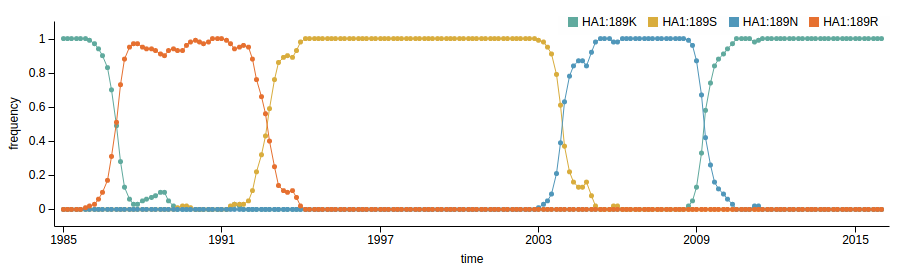
- Influenza virus evolves to avoid human immunity
- Vaccines need frequent updates
Predicting evolution
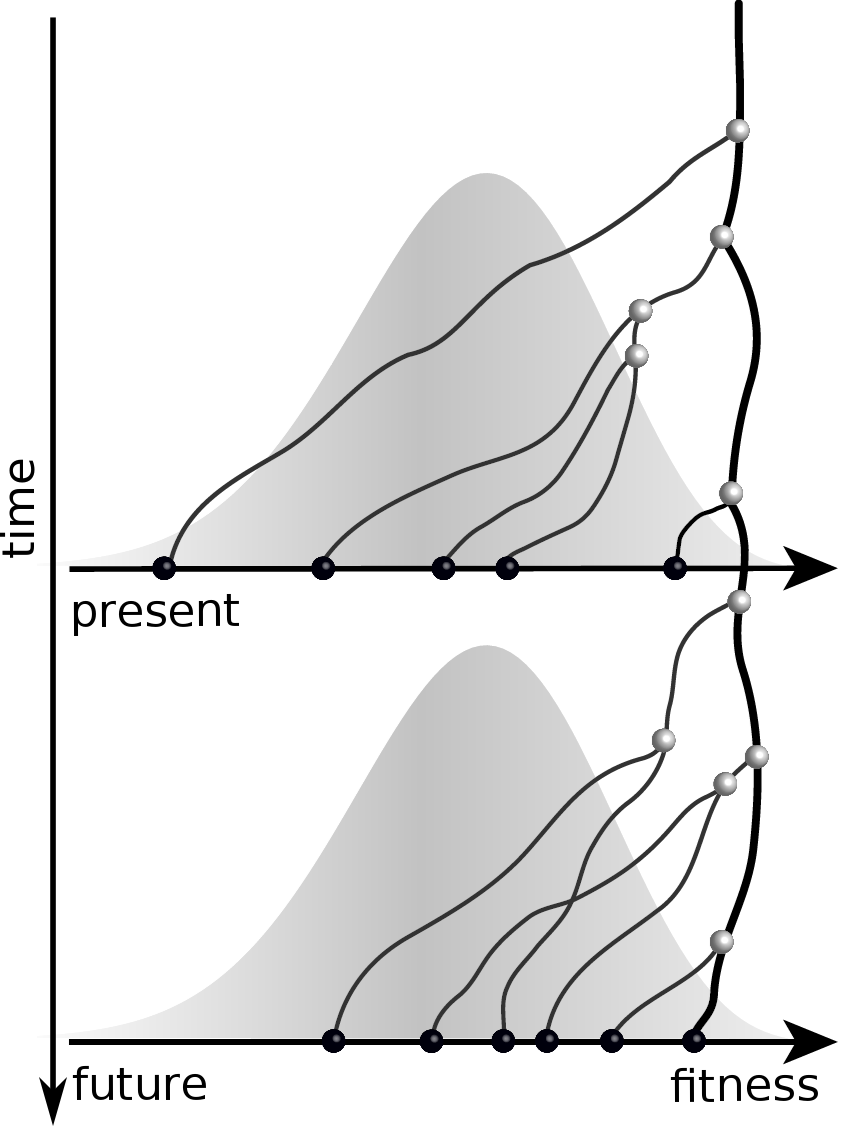
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
Validate on simulation data
- simulate evolution
- sample sequences
- reconstruct trees
- infer fitness
- predict ancestor of future
- compare to truth
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
Summary
- RNA virus evolution can be observed directly
- Extensive reversion to preferred amino acid sequence
- Rapidly adapting population require new population genetic models
- Those model can be used to infer fit clades
- Future influenza population can be anticipated
- Automated real-time analysis can help fight the spread of disease
HIV acknowledgments







- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
nextstrain.org





- Trevor Bedford
- Colin Megill
- Pavel Sagulenko
- Sidney Bell
- James Hadfield
- Wei Ding
