Virus evolution and the predictability of next year's flu
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201802_TUM.html
Evolution of HIV
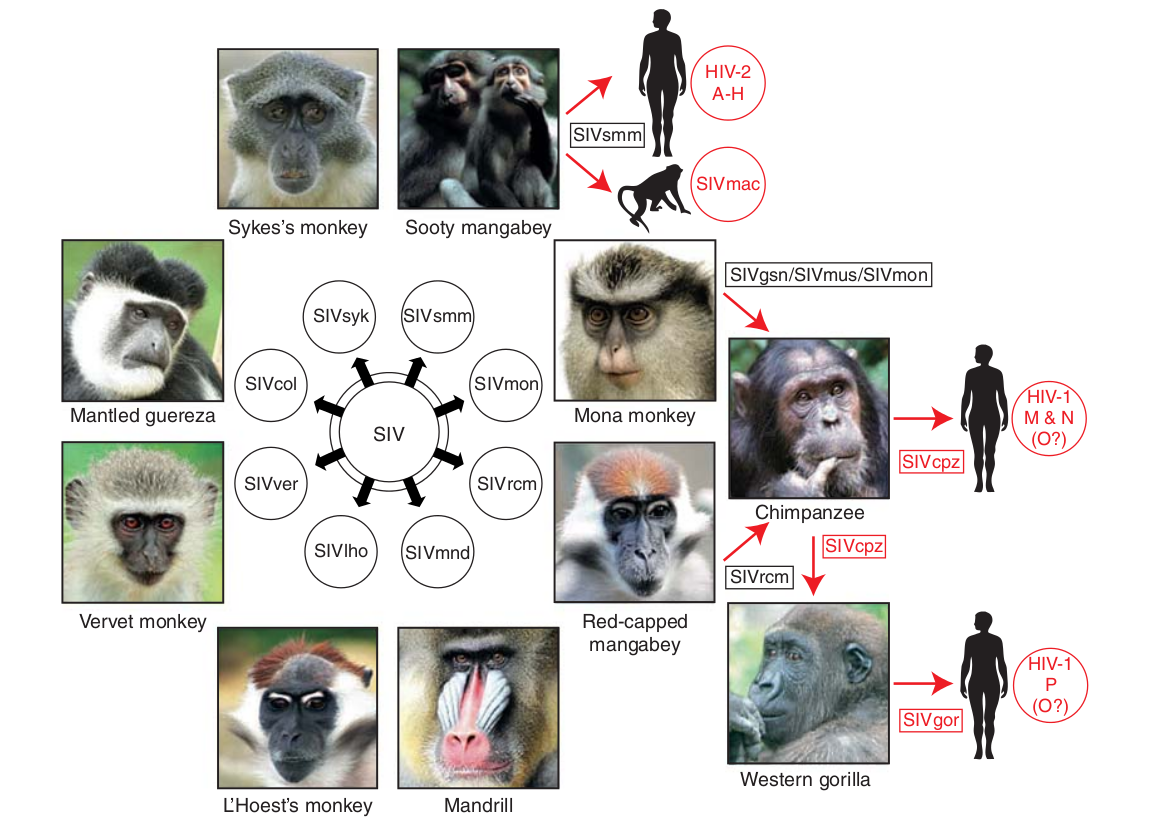
- Chimp → human transmission around 1900 gave rise to HIV-1 group M
- ~100 million infected people since
- subtypes differ at 10-20% of their genome
- HIV-1 evolves ~0.1% per year
HIV infection
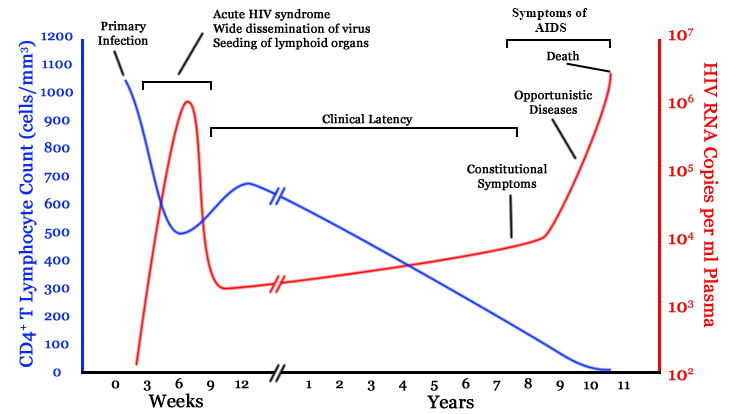

- $10^8$ cells are infected every day
- the virus repeatedly escapes immune recognition
- integrates into T-cells as
latent provirus
{kind=link}
HIV-1 evolution within one individual

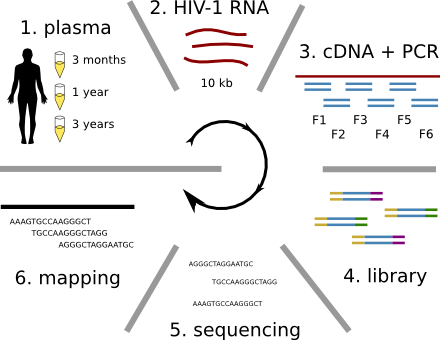

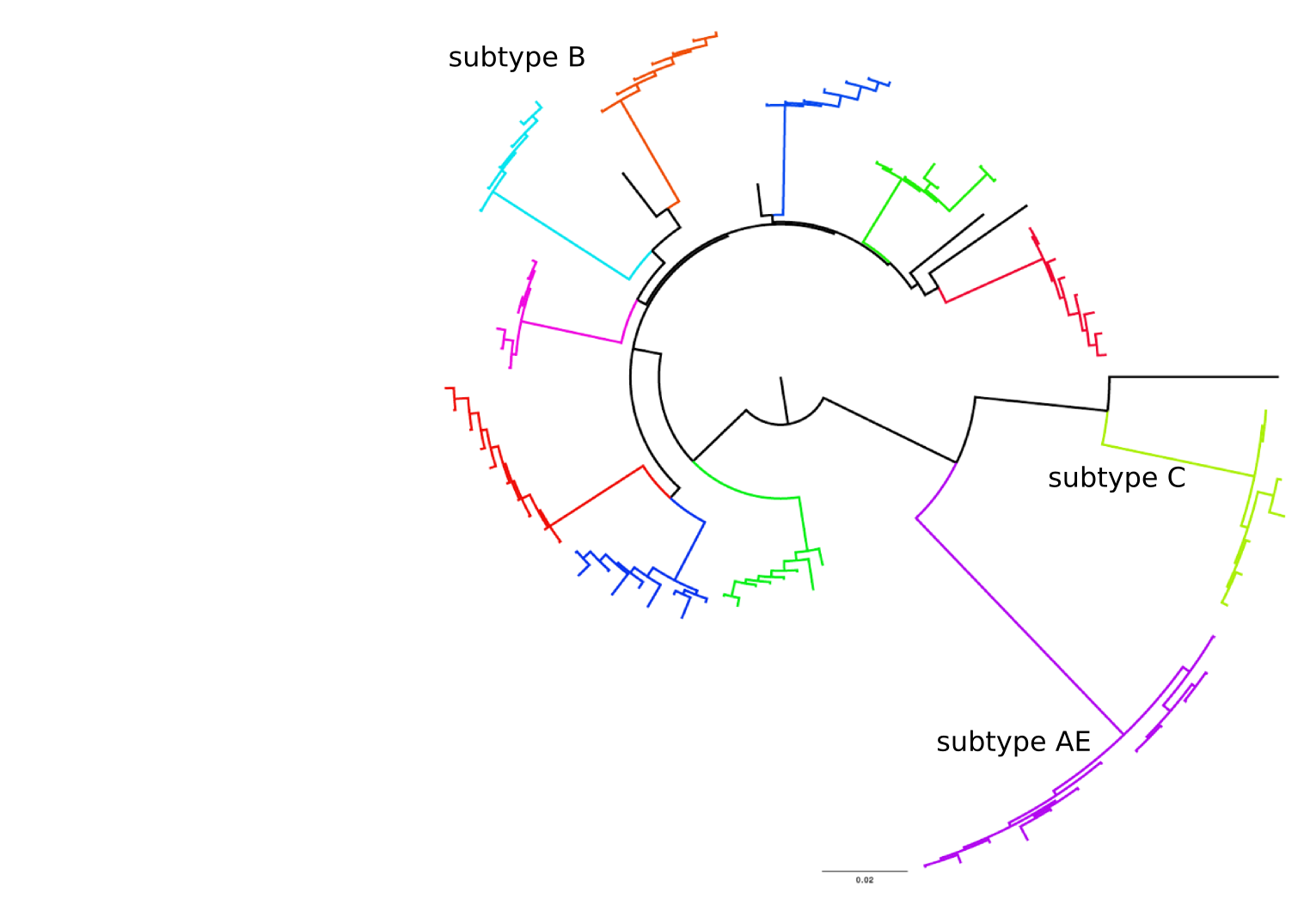
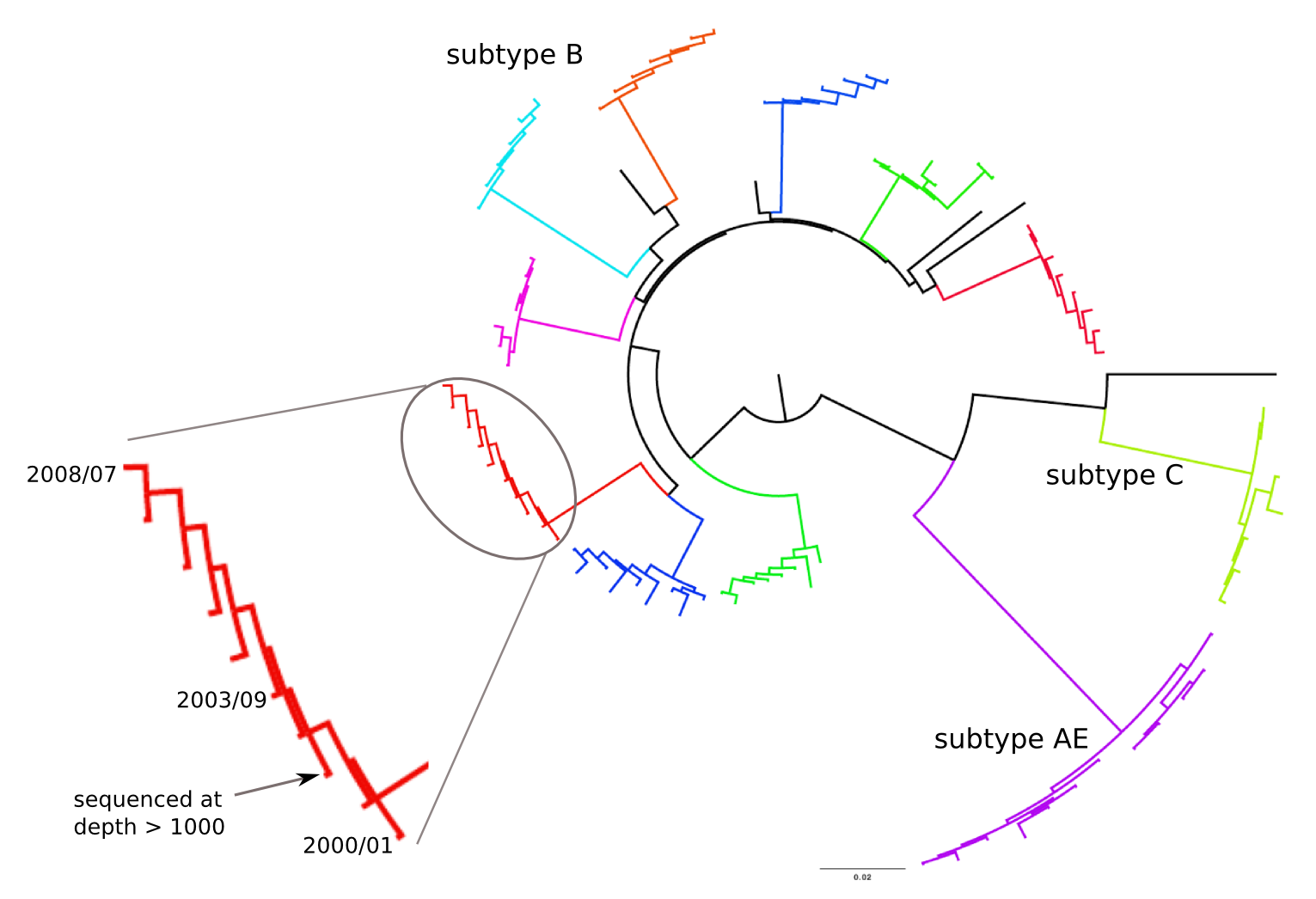
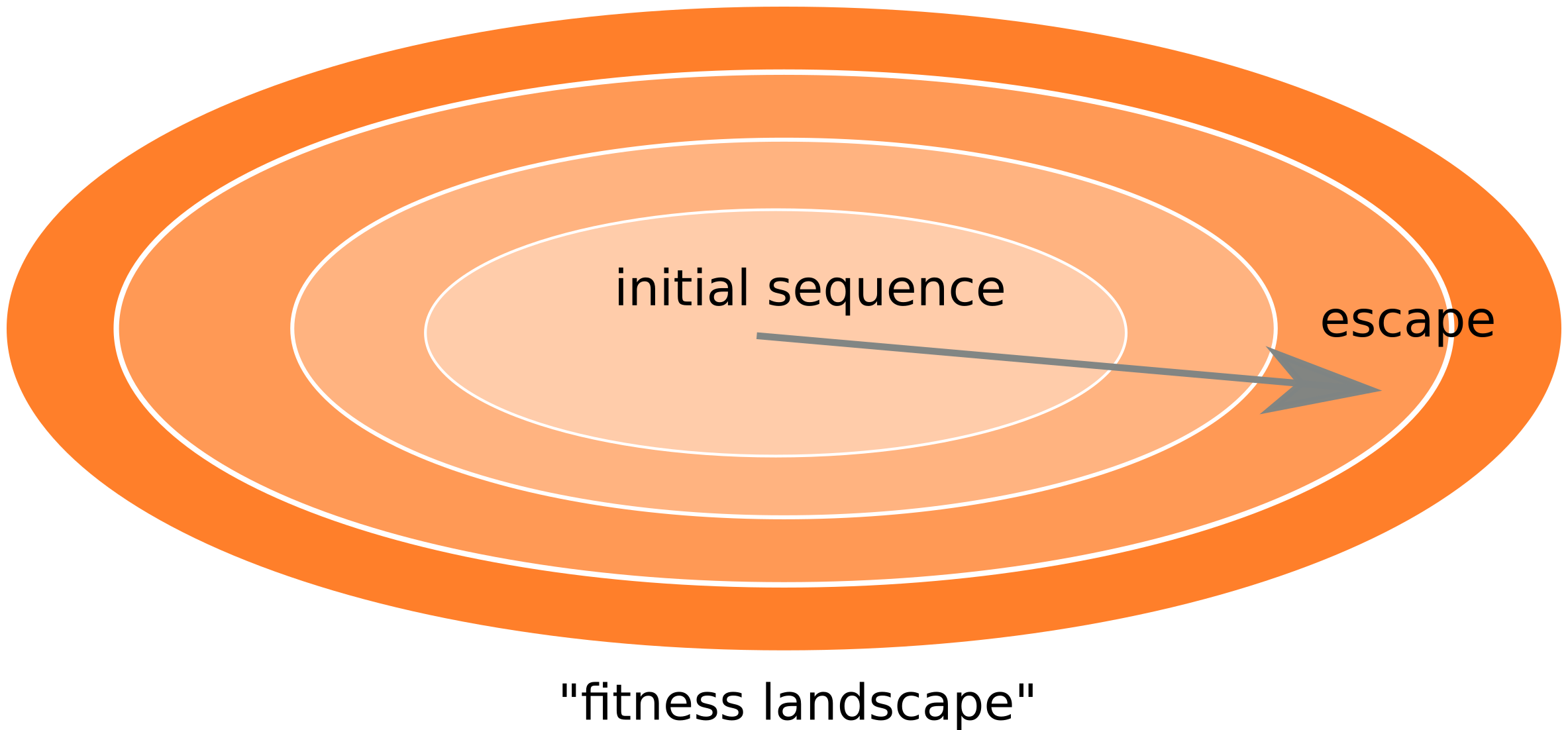
Frequent reversion of previously beneficial mutations
- HIV escapes immune systems
- most mutations are costly
- humans selects for different mutations
- compensation or reversion?
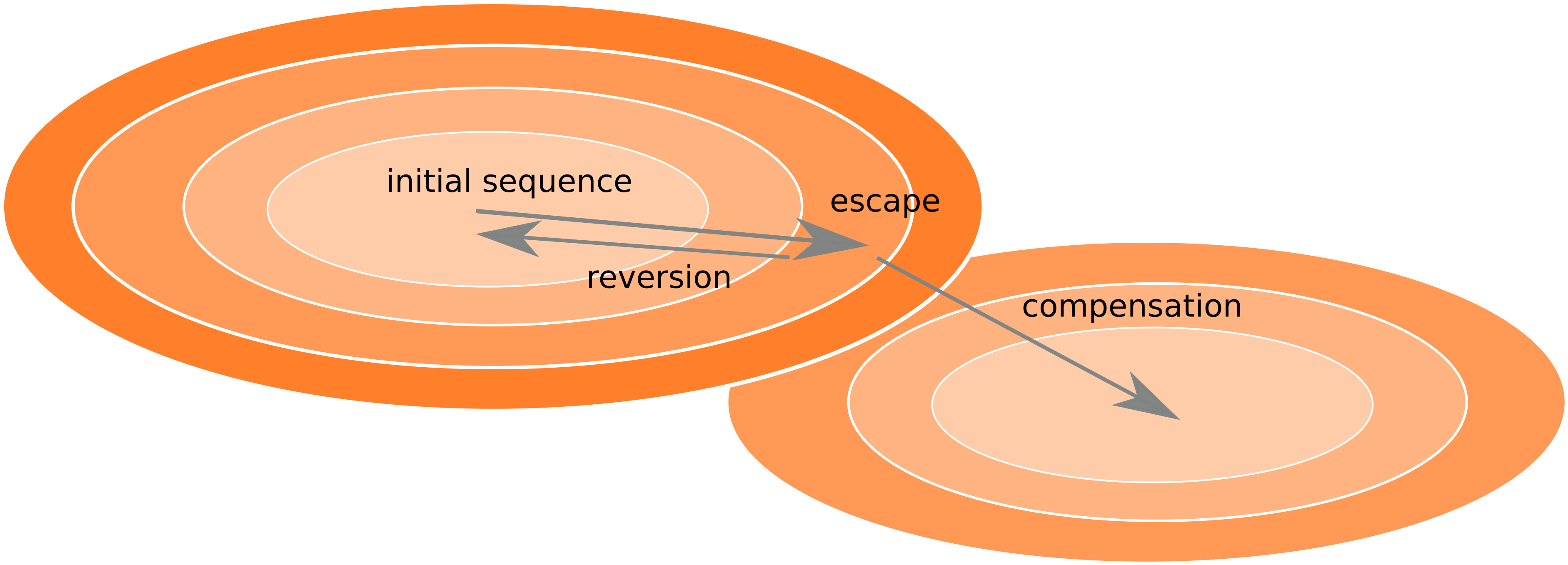
Theoretical framework for virus evolution -- population genetics




evolutionary processes ↔ trees ↔ genetic diversity
Neutral models and beyond
Neutral models
- all individuals are identical → same offspring distribution
- Kingman coalesence and diffusion theory are dual descriptions
- everything is easy to calculate
- perturbations like background selection can be included
But: neutral models not suitable for RNA viruses!
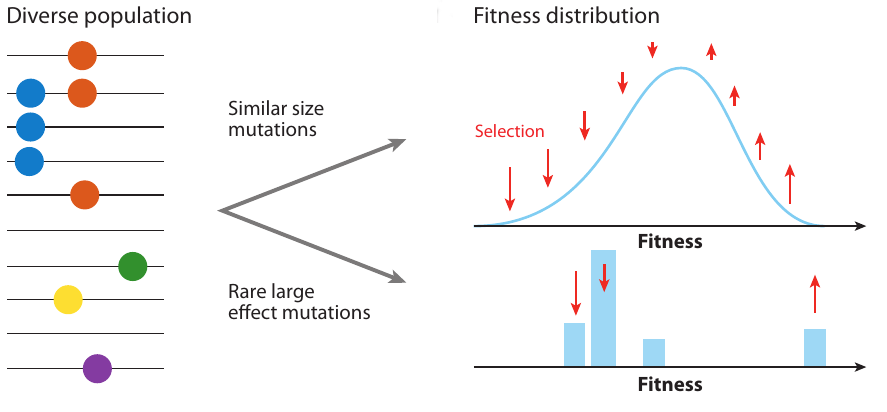
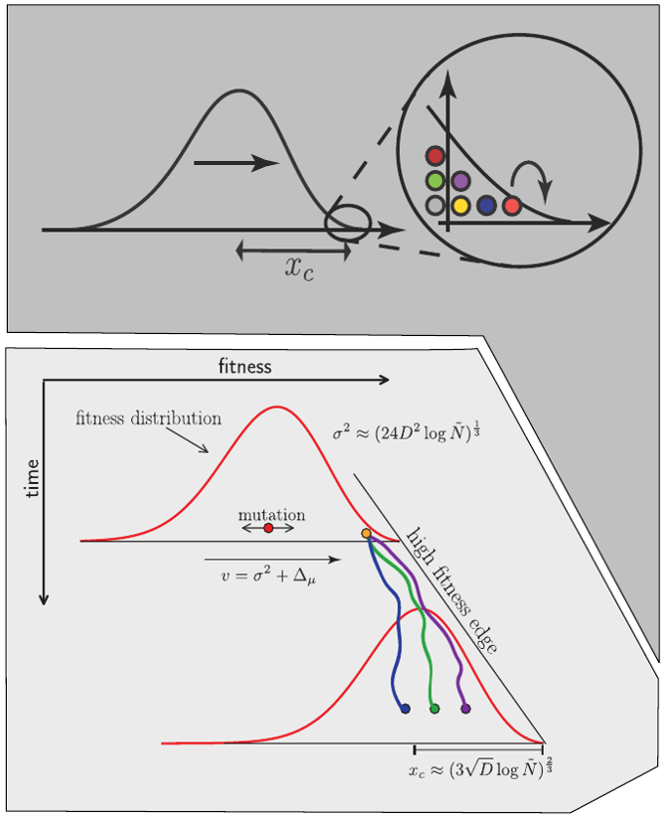
Clonal interference and traveling waves
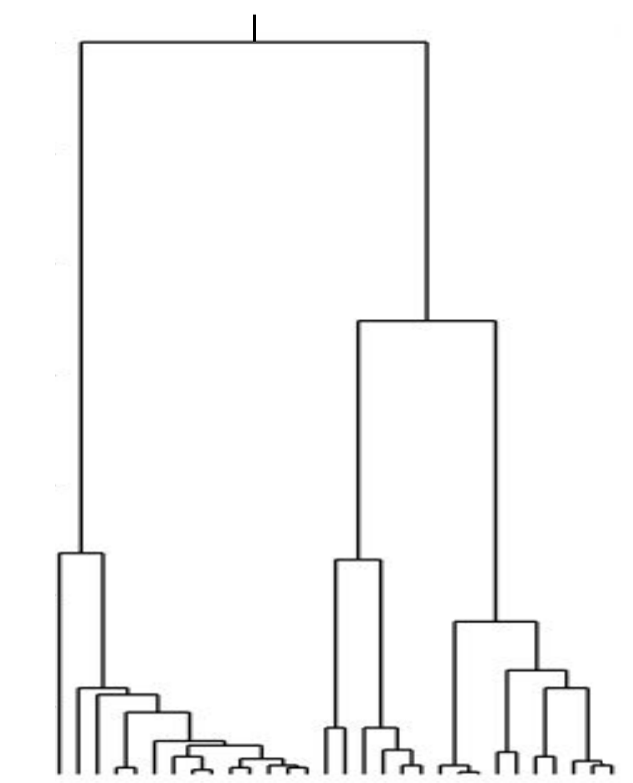
Neutral/Kingman coalescent
strong selection
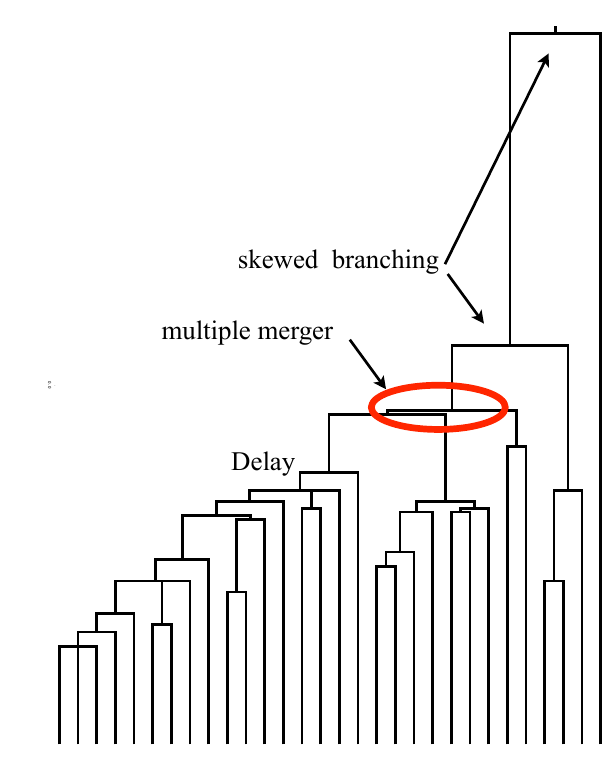
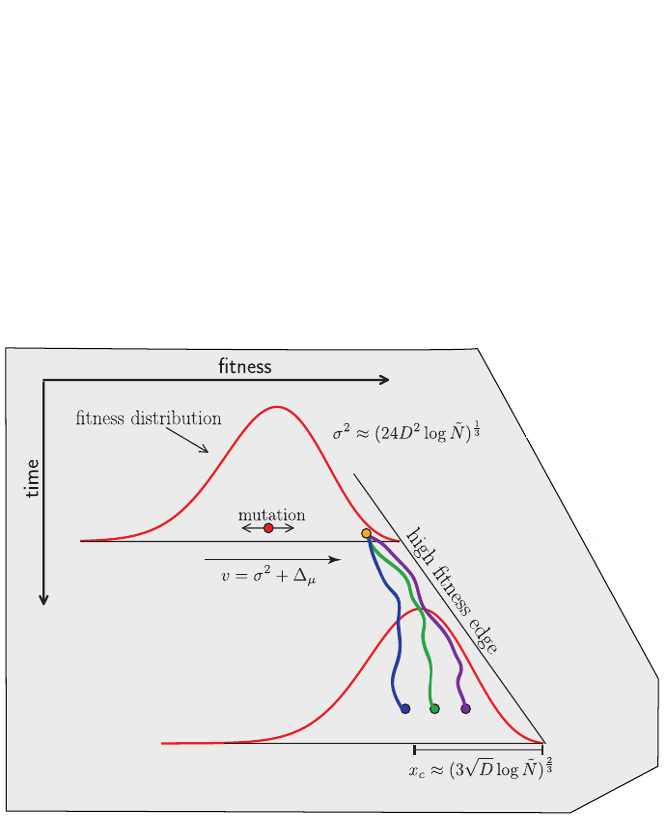
Bolthausen-Sznitman Coalescent
U-shaped polarized site frequency spectra
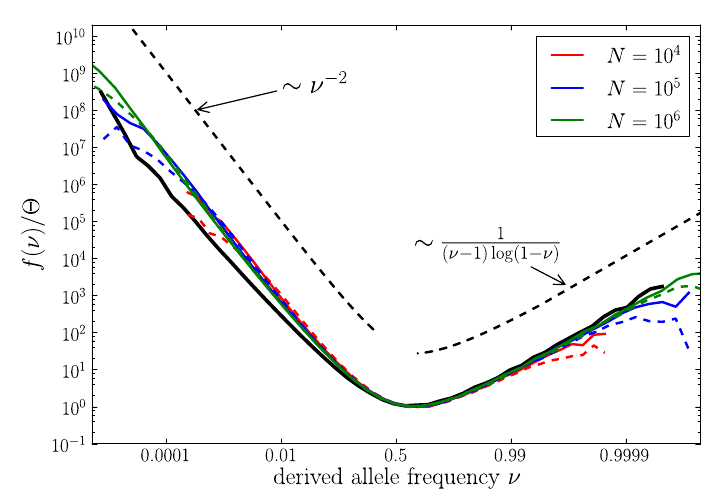

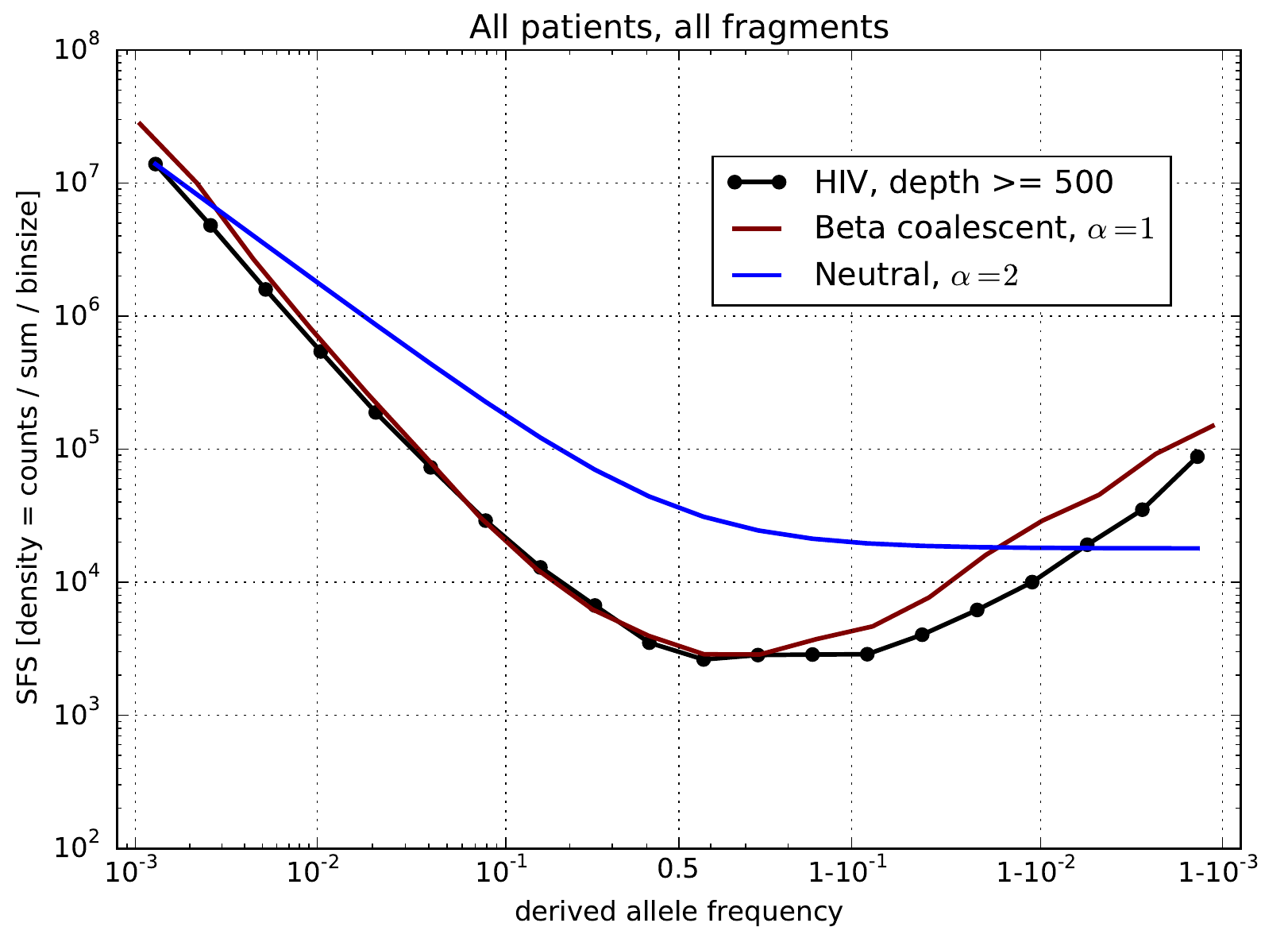 Zanini et al, eLife, 2015
Zanini et al, eLife, 2015
Bursts in a tree ↔ high fitness genotypes
Can we read fitness of a tree?
Human seasonal influenza viruses
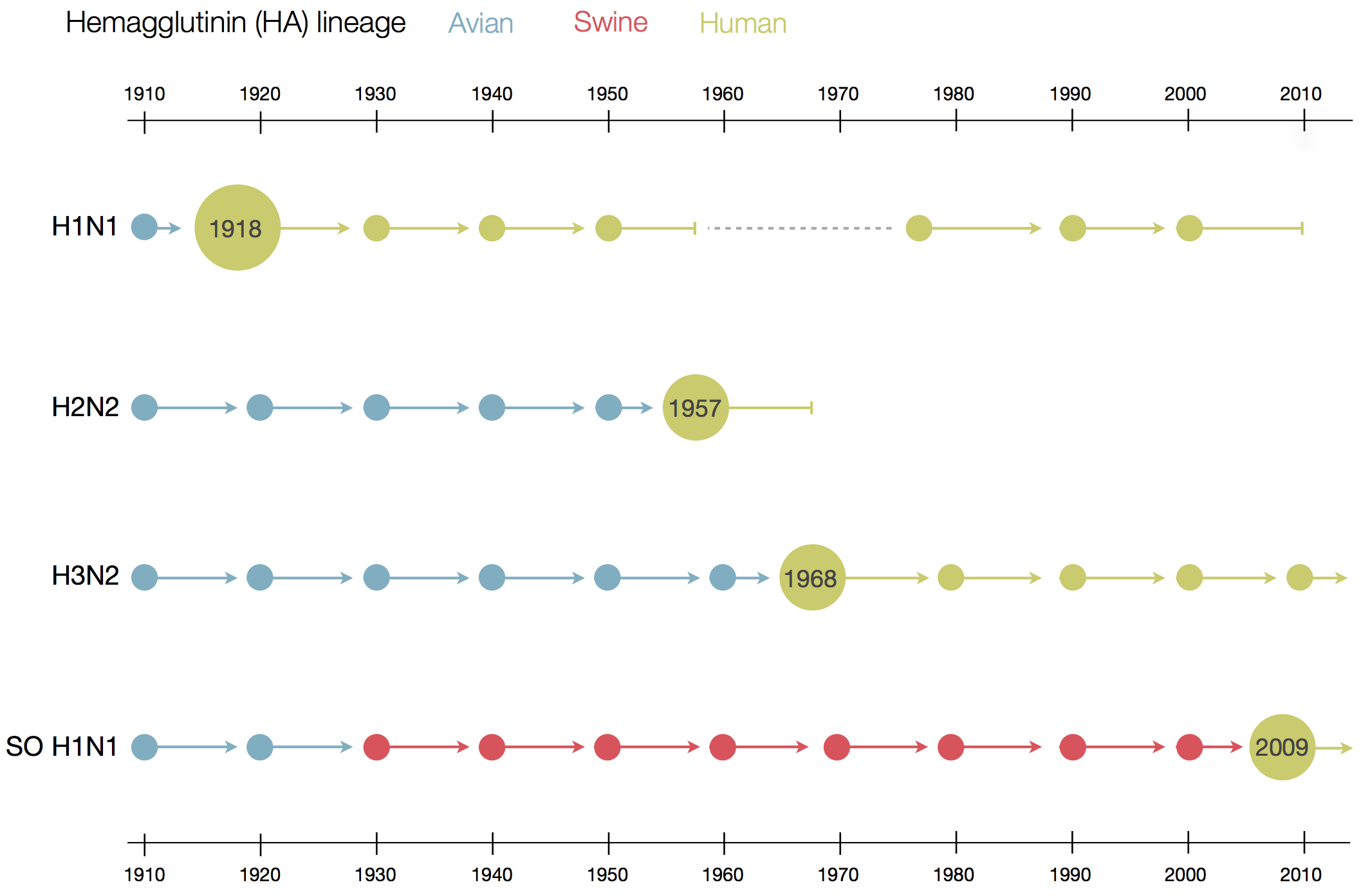
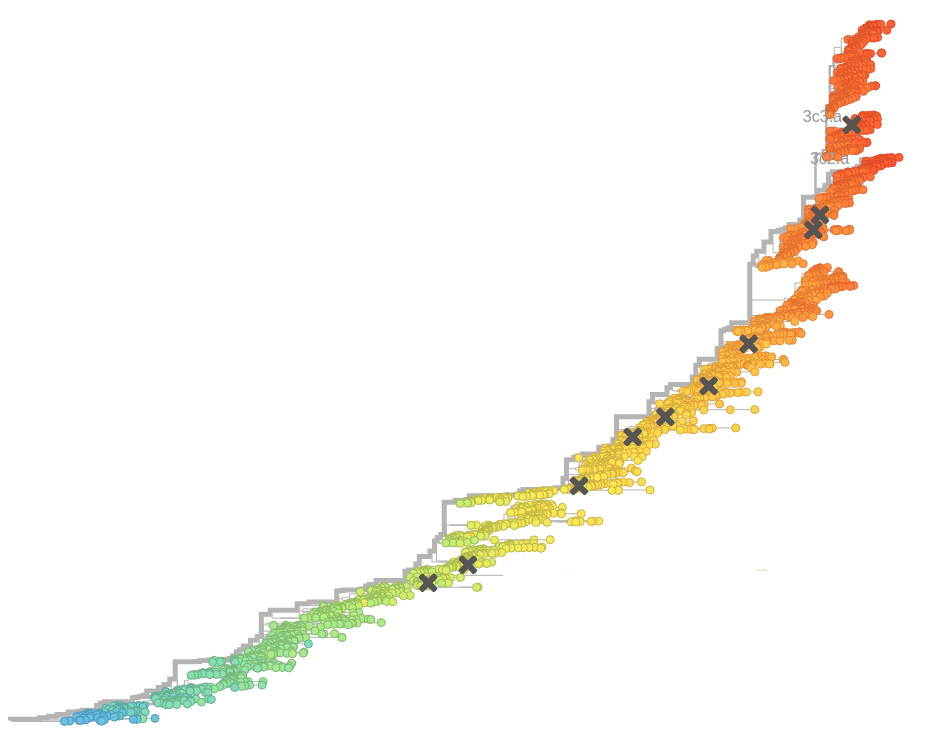
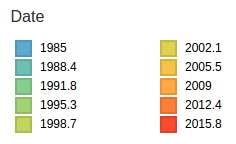
- Influenza virus evolves to avoid human immunity
- Vaccines need frequent updates
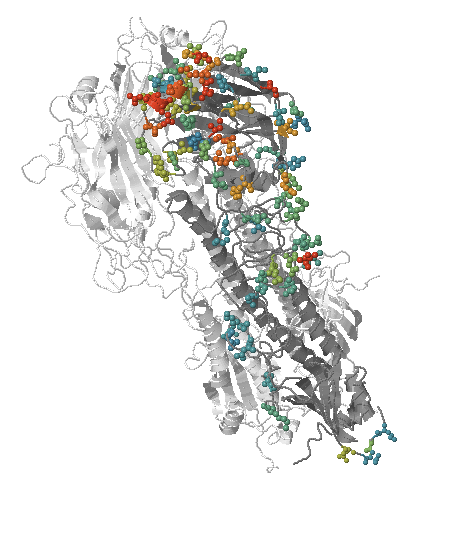
Predicting evolution
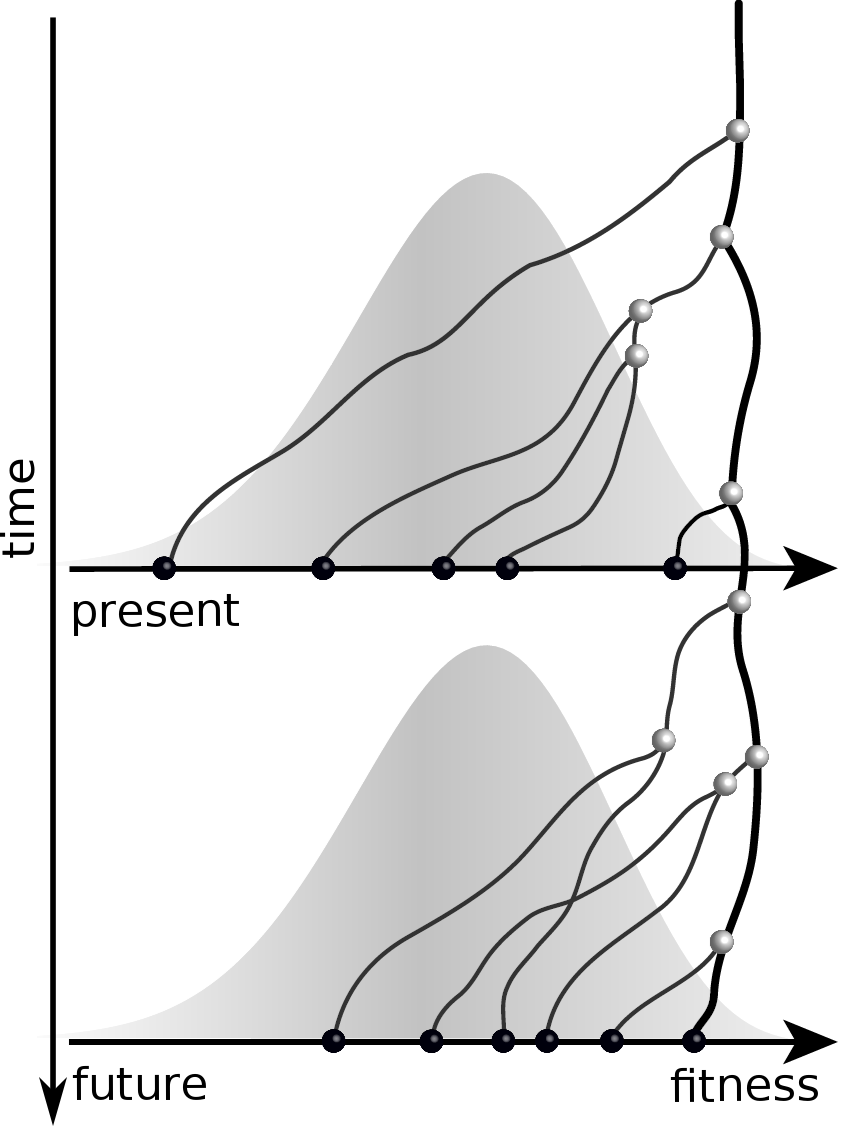
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
Validate on simulation data
- simulate evolution
- sample sequences
- reconstruct trees
- infer fitness
- predict ancestor of future
- compare to truth
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
nextstrain.org
Summary
- RNA virus evolution can be observed directly
- Extensive reversion to preferred amino acid sequence
- Rapidly adapting population require new population genetic models
- Those model can be used to infer fit clades
- Future influenza population can be anticipated
- Automated real-time analysis can help fight the spread of disease
HIV acknowledgments
- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
nextstrain.org
- Trevor Bedford
- Colin Megill
- Pavel Sagulenko
- Wei Ding
- Sidney Bell
- James Hadfield
