Real time analysis and visualization of RNA virus evolution
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201803_XLAB.html
Viruses
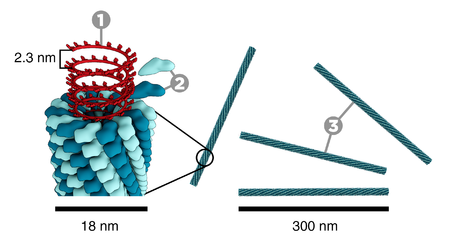 tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
bacteria phage (adenosine, wikipedia)
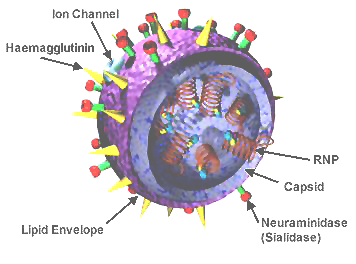
influenza virus wikipedia

human immunodeficiency virus wikipedia
- rely on host to replicate
- little more than genome + capsid
- genomes typically 5-200k bases (+exceptions)
- many infectious diseases are caused by viruses
- very important function in microbial eco-systems
- most abundant organisms on earth $\sim 10^{31}$
Some viruses evolve a million times faster than animals
Animal haemoglobin

HIV protein

Development of sequencing technologies
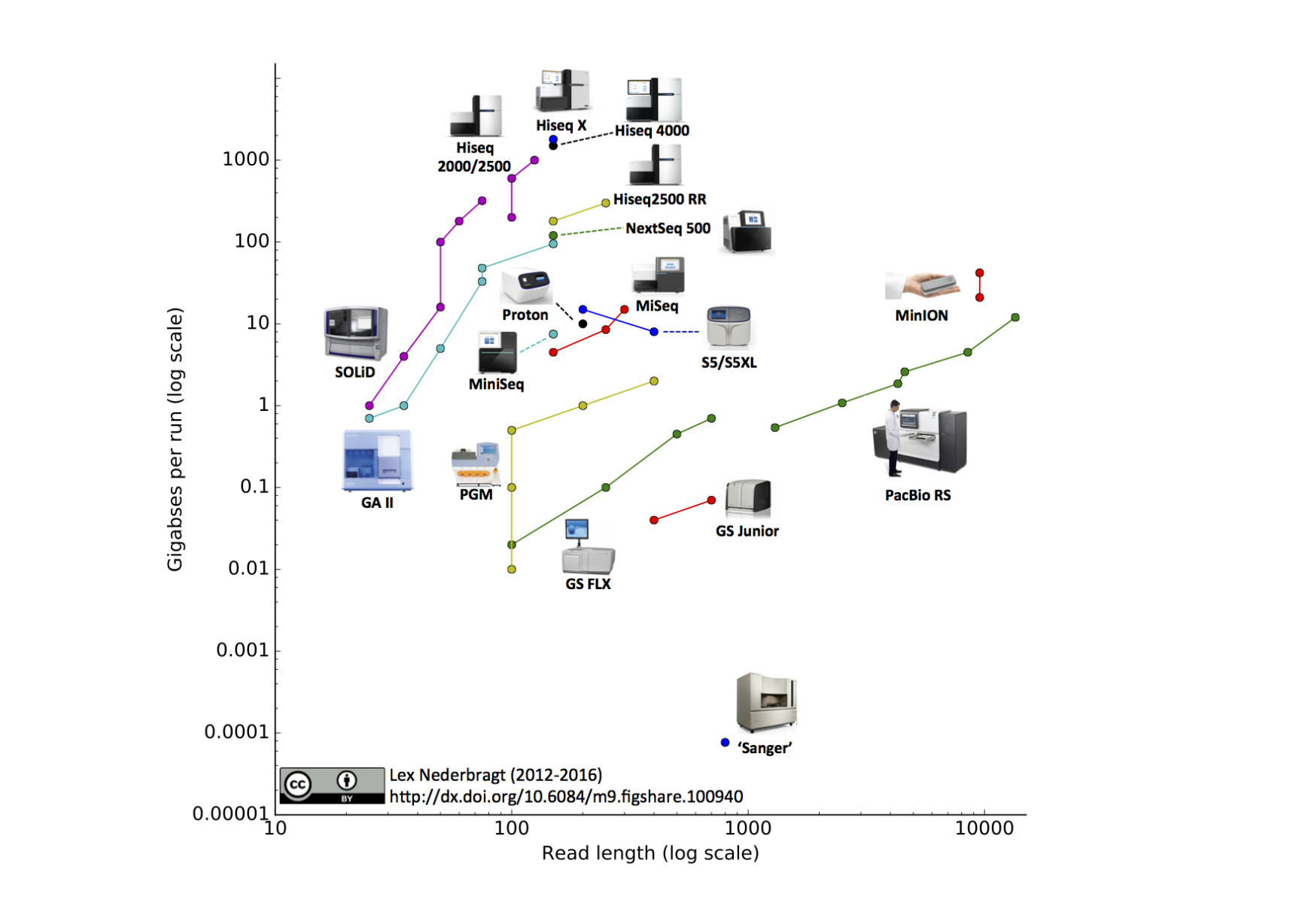
We can now sequence...
- thousands of bacterial isolates
- thousands of single cells
- populations of viruses, bacteria or flies
- diverse ecosystems
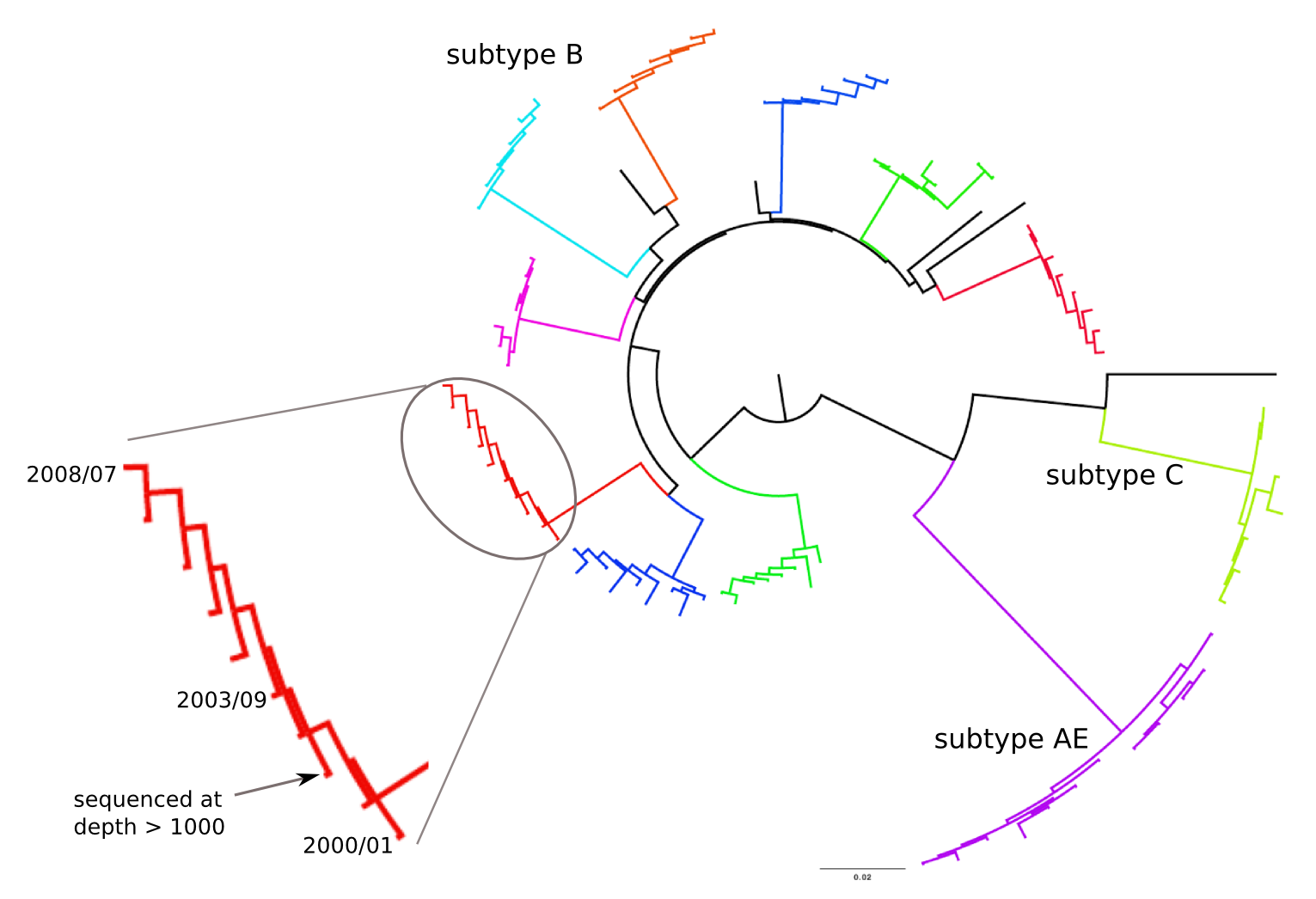
Evolution of HIV
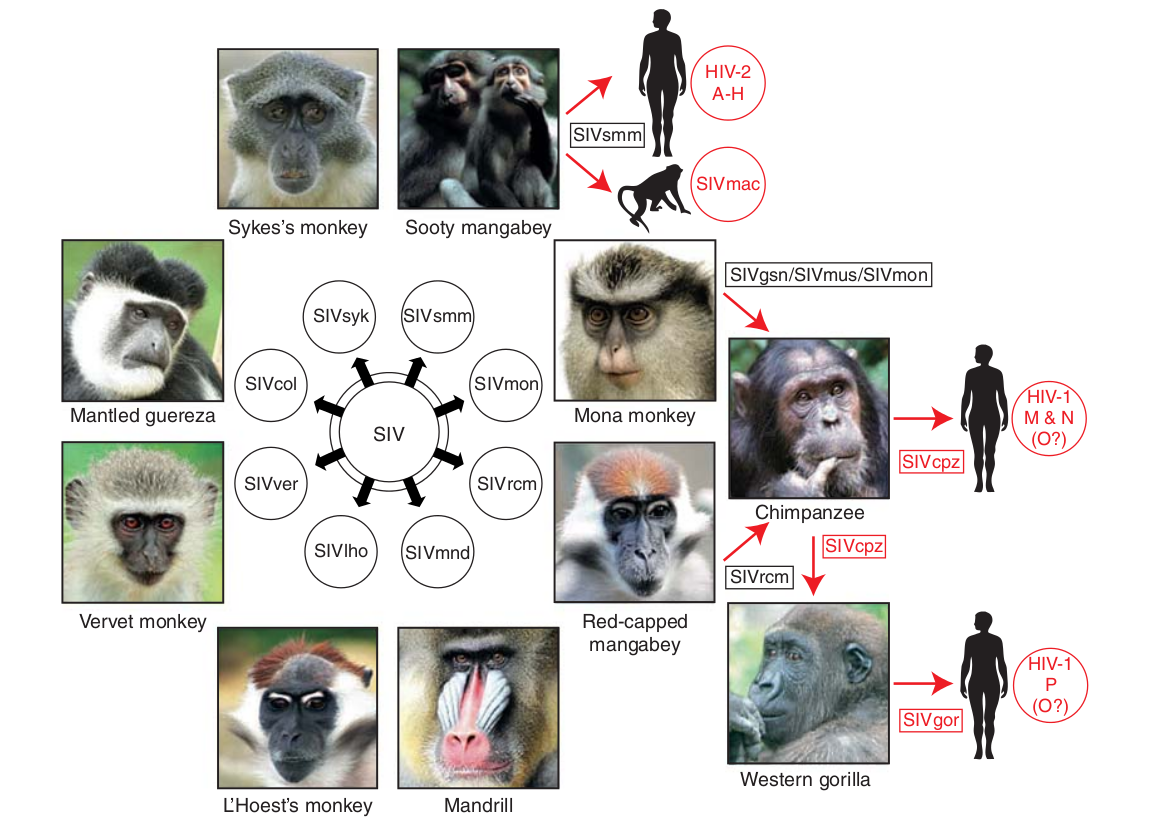
- Chimp → human transmission around 1900 gave rise to HIV-1 group M
- ~100 million infected people since
- subtypes differ at 10-20% of their genome
- HIV-1 evolves ~0.1% per year
HIV infection
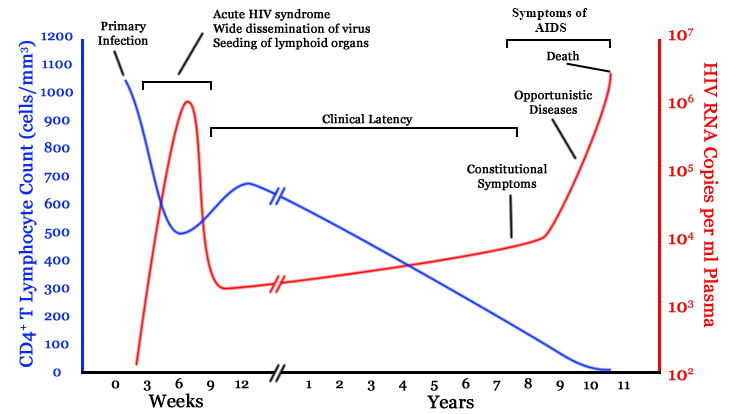

- $10^8$ cells are infected every day
- the virus repeatedly escapes immune recognition
- integrates into T-cells as
latent provirus
{kind=link}
HIV-1 evolution within one individual

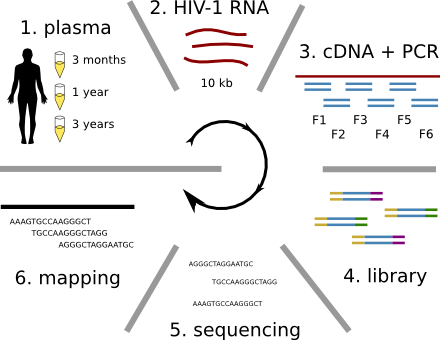

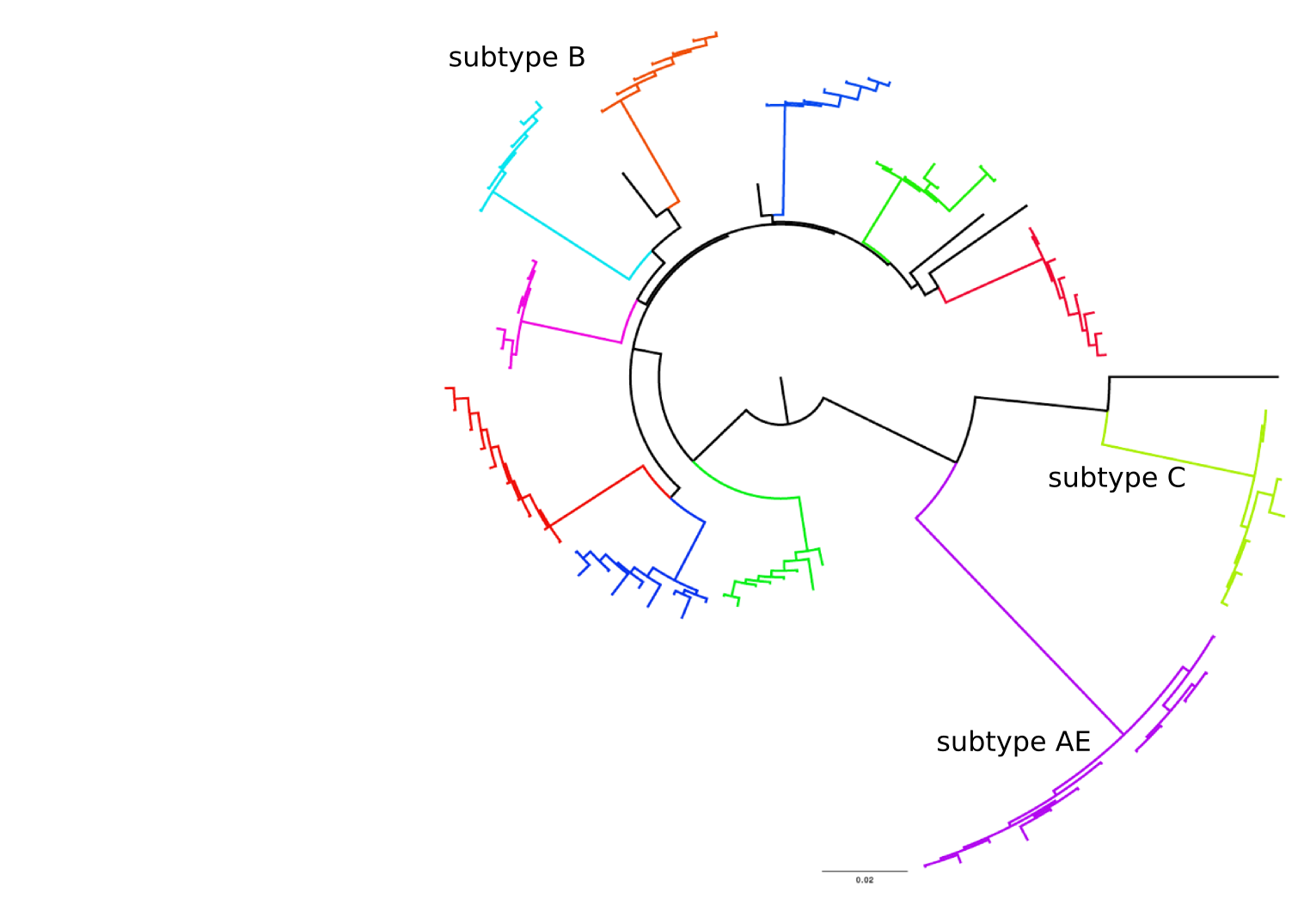
Immune escape in early HIV infection
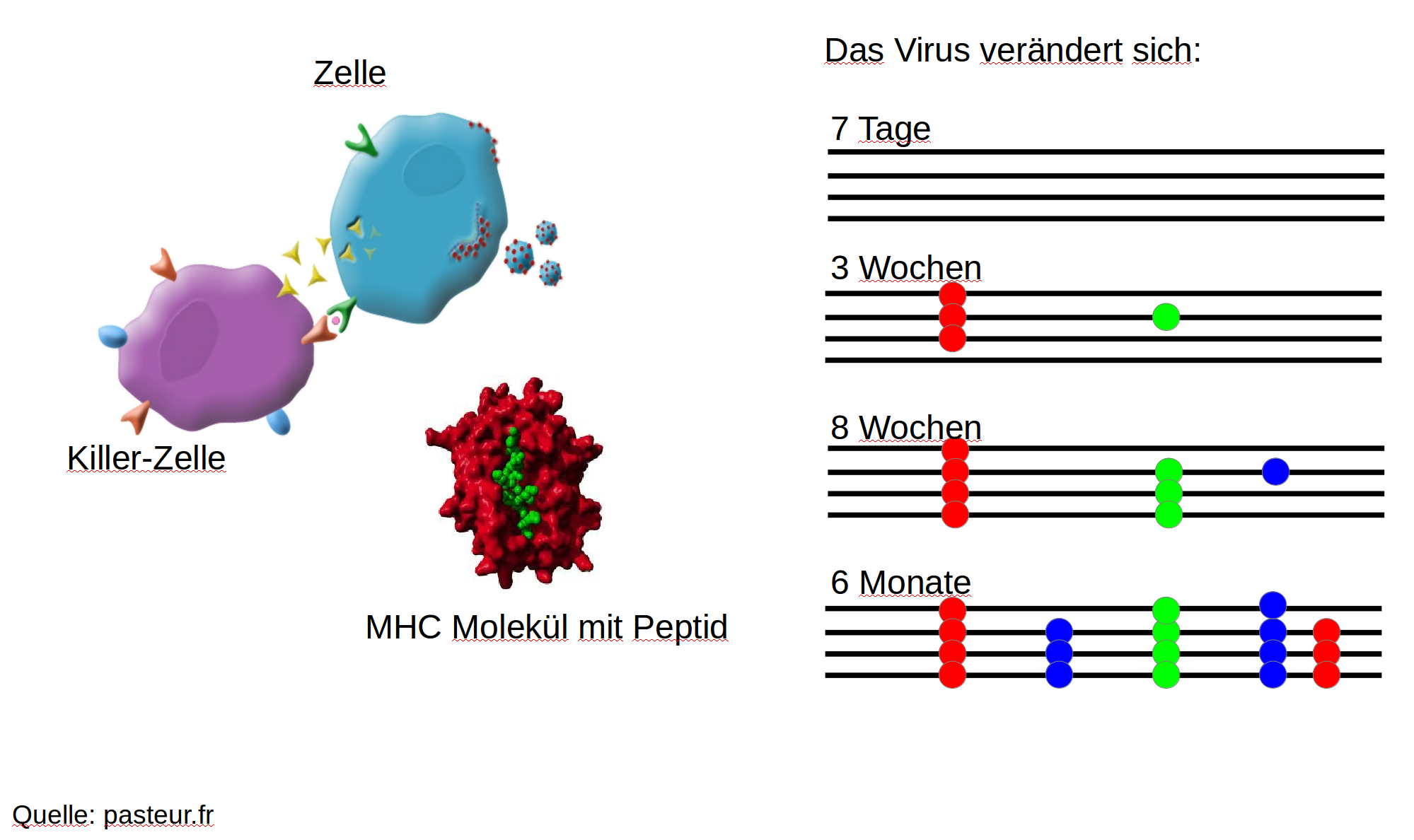
Immune escape in early HIV infection
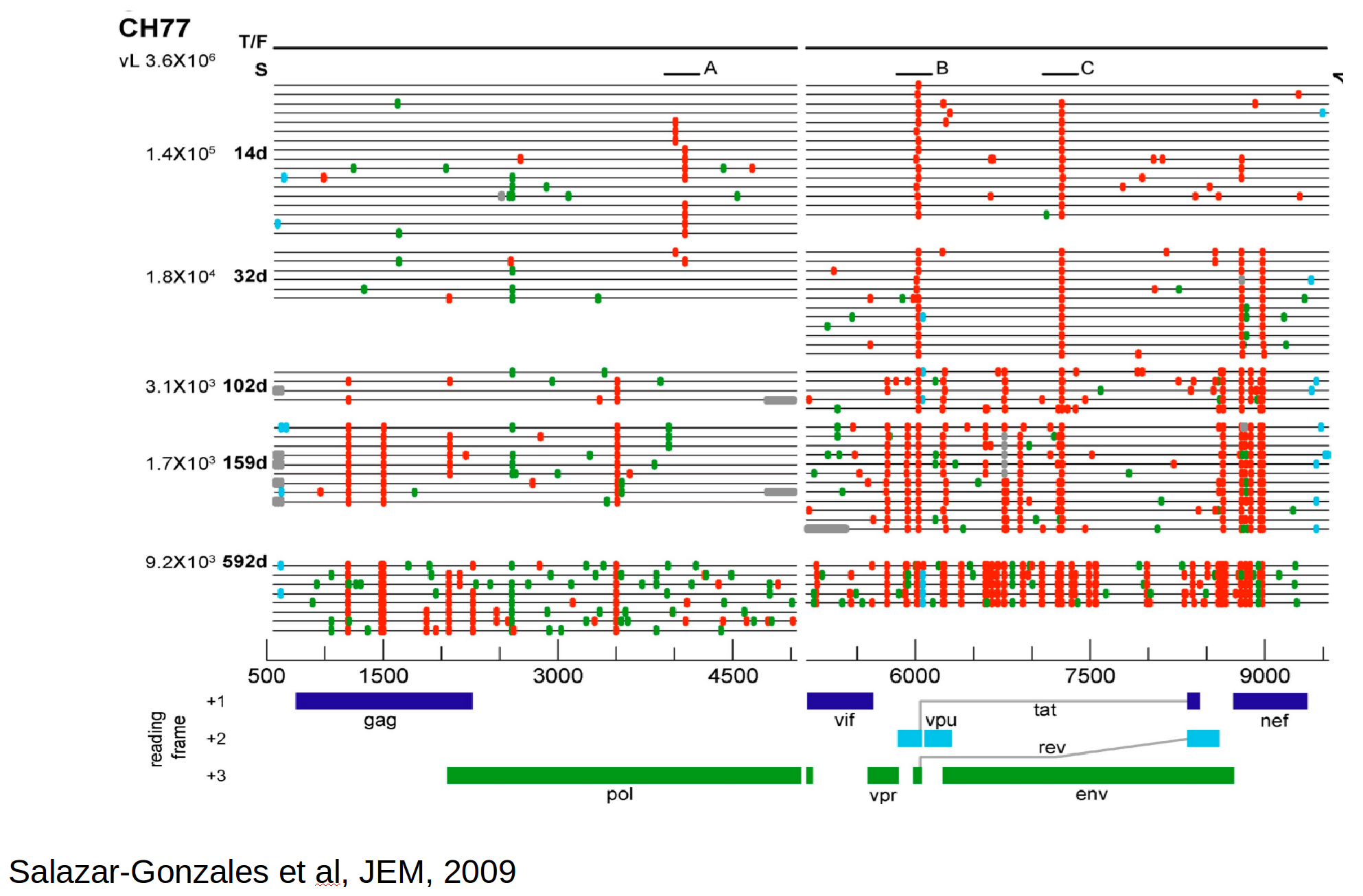
Population genetics & evolutionary dynamics




evolutionary processes ↔ trees ↔ genetic diversity
Selective sweeps
- Viruses carrying a beneficial mutation have more offspring: on average $1+s$ instead of $1$
- $s$ is called selection coefficient
- Fraction $x$ of viruses carrying the mutation changes as $$x(t+1) = \frac{(1+s)x(t)}{(1+s)x(t) + (1-x(t))}$$
- In continuous time → logistic differential equation: $$\frac{dx}{dt} = sx(1-x) \Rightarrow x(t) = \frac{e^{s(t-t_0)}}{1+ e^{s(t-t_0)}}$$
The rate of sequence evolution in HIV
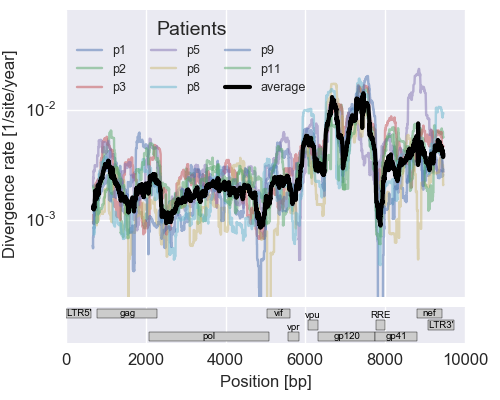
Mutation rates and diversity and neutral sites
 Zanini et al, Virus Evolution, 2017
Zanini et al, Virus Evolution, 2017
Inference of fitness costs
- mutation away from preferred state with rate $\mu$
- selection against non-preferred state with strength $s$
- variant frequency dynamics: $\frac{d x}{dt} = \mu -s x $
- equilibrium frequency: $\bar{x} = \mu/s $
- fitness cost: $s = \mu/\bar{x}$
Fitness landscape of HIV-1
Zanini et al, Virus Evolution, 2017Selection on RNA structures and regulatory sites
Zanini et al, Virus Evolution, 2017The distribution of fitness costs
Zanini et al, Virus Evolution, 2017Sequences record the spread of pathogens
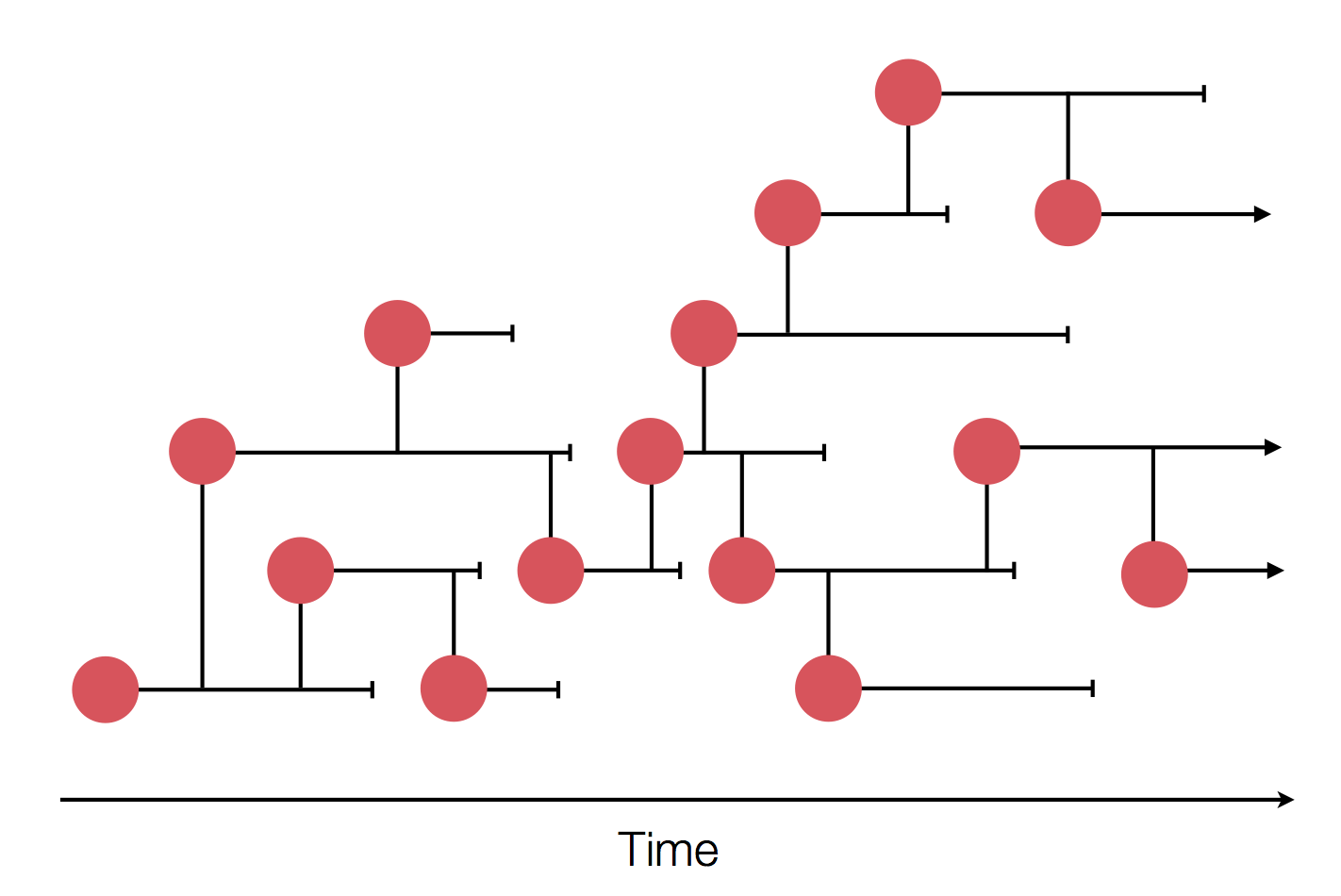
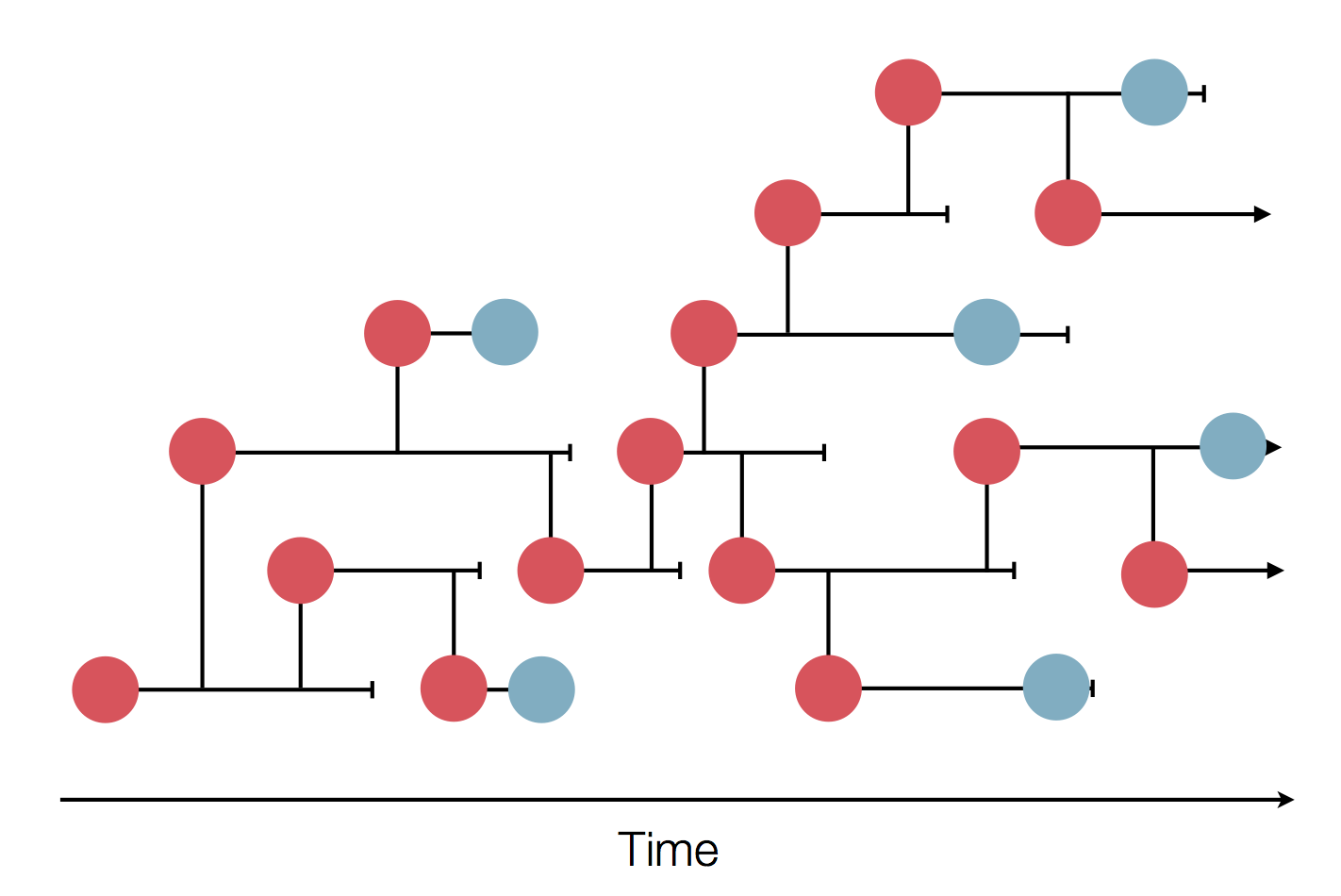
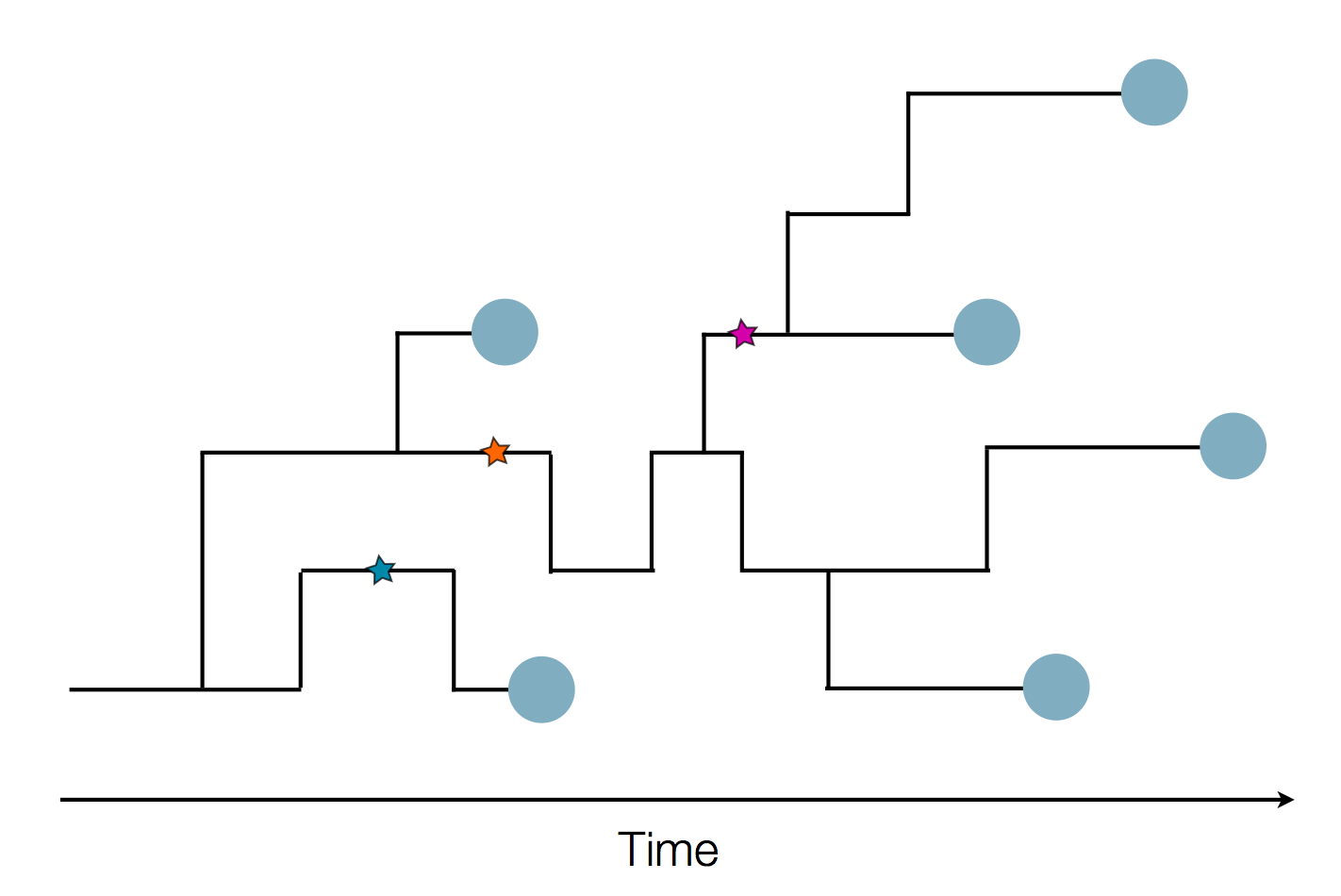
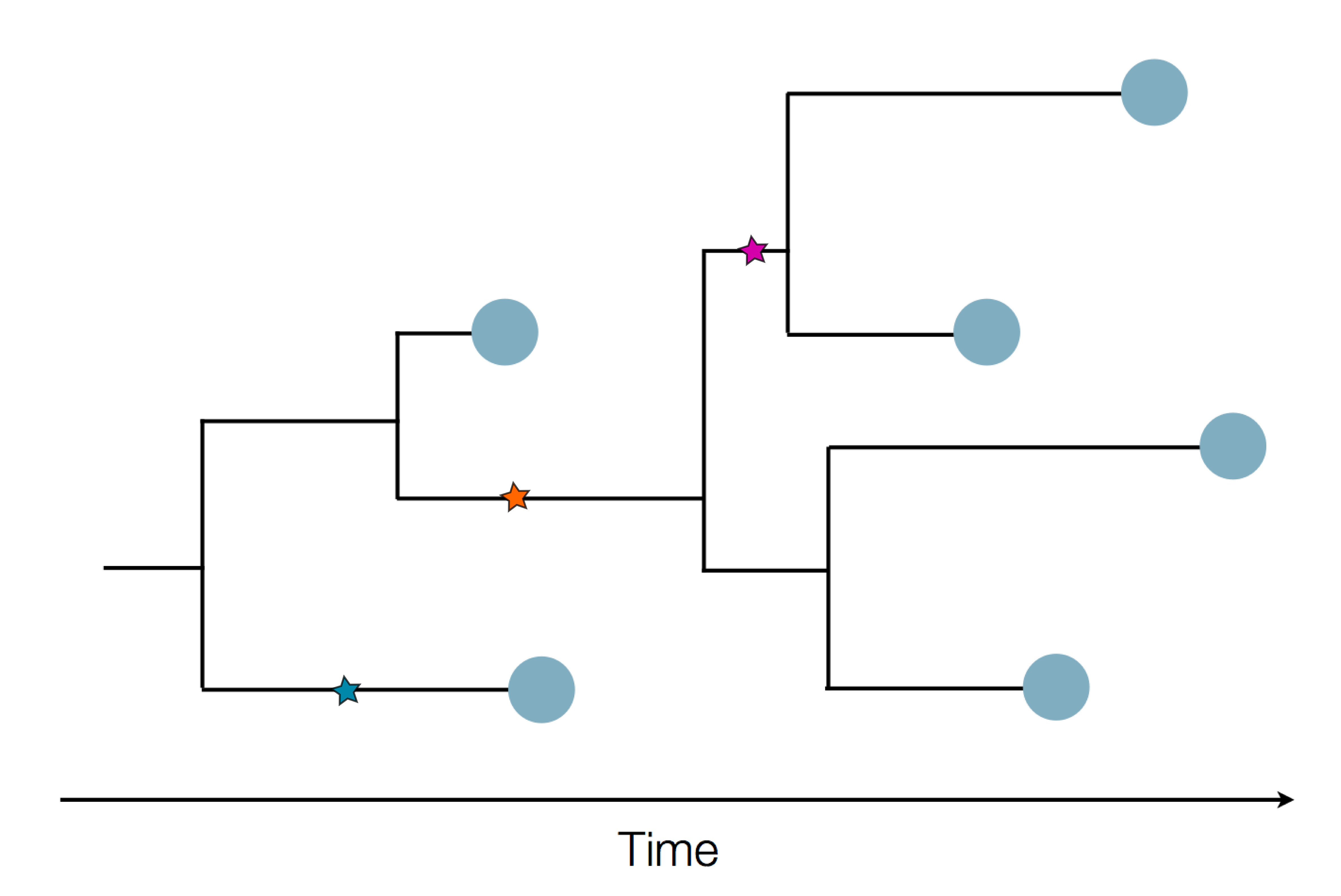
The resolution is limited by the number of mutations!
Influenza virus genome - 8 segments
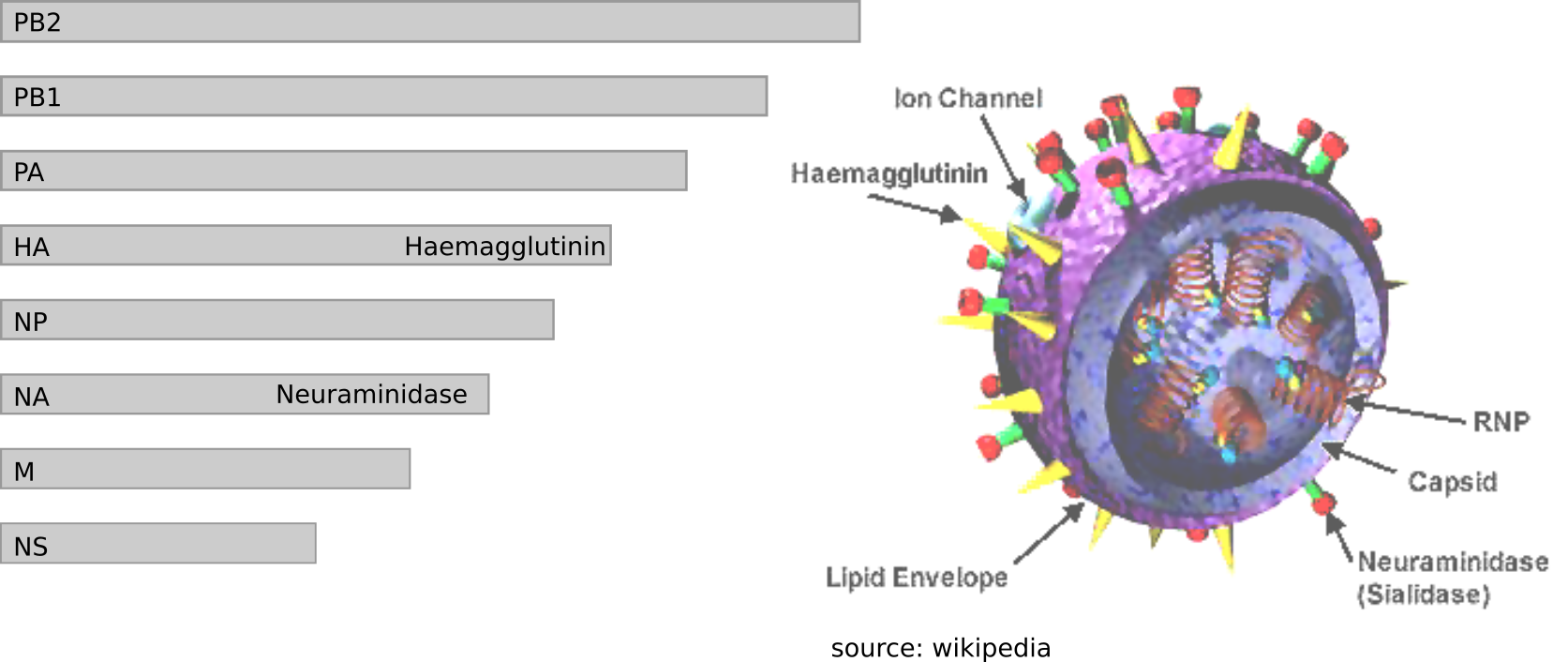
Zika virus genome $\sim 10000$ bases

Ebola virus genome $\sim 20000$ bases

Many RNA viruses pick up one mutation every 2-4 weeks!
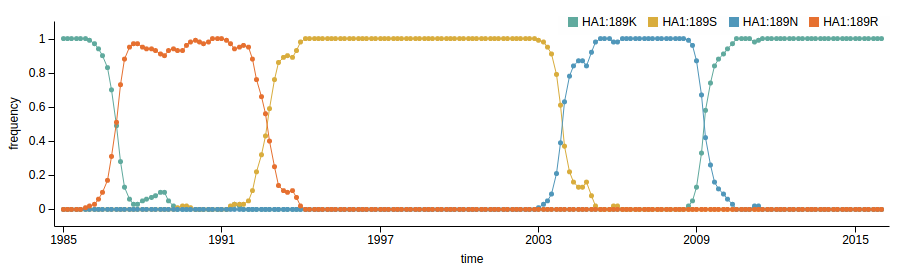
Human seasonal influenza viruses
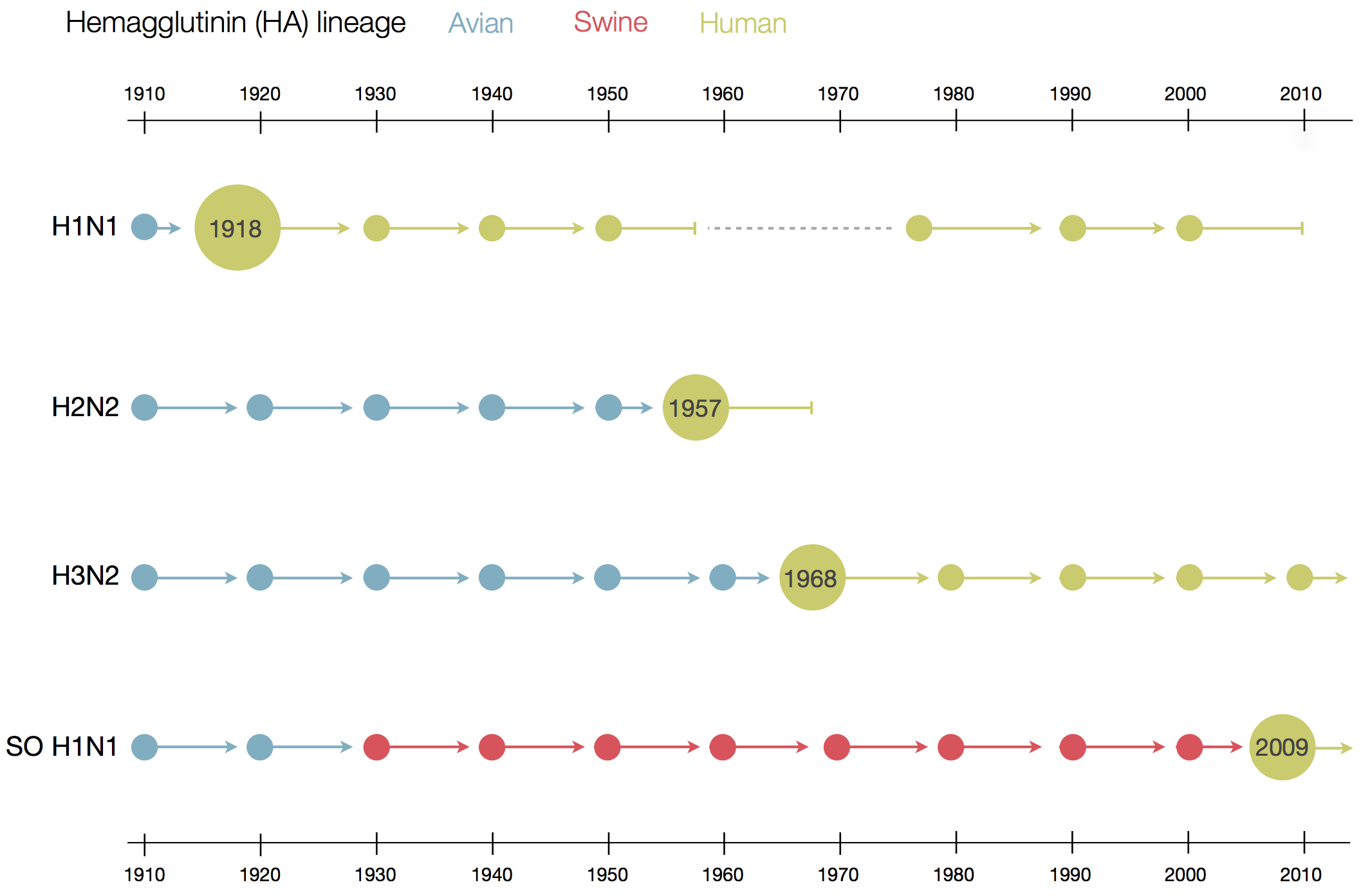
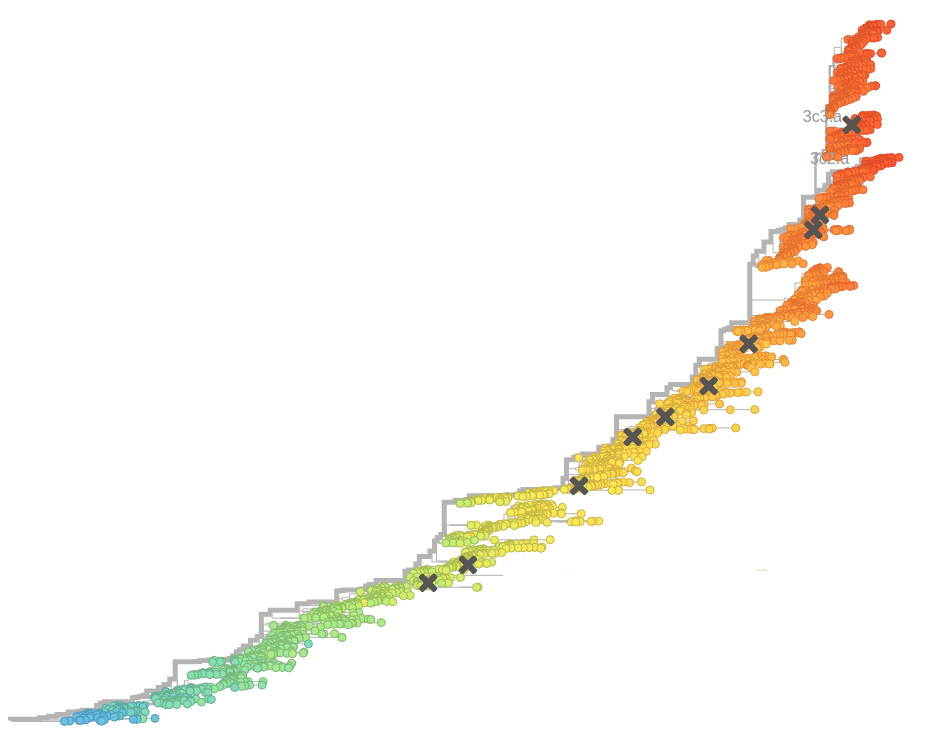
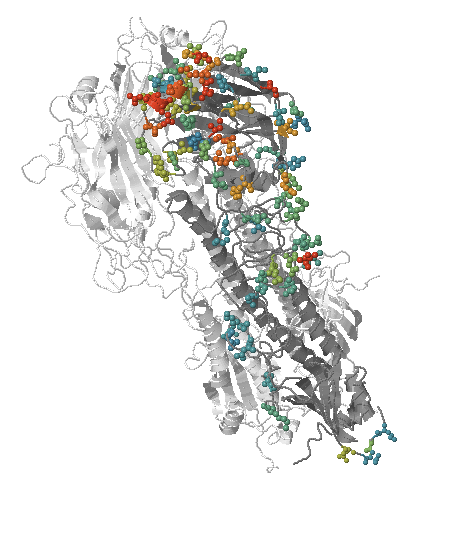
- Influenza viruses evolve to avoid human immunity
- Vaccines need frequent updates
nextflu.org
joint work with Trevor Bedford & his lab
code at github.com/blab/nextflu
Predicting evolution
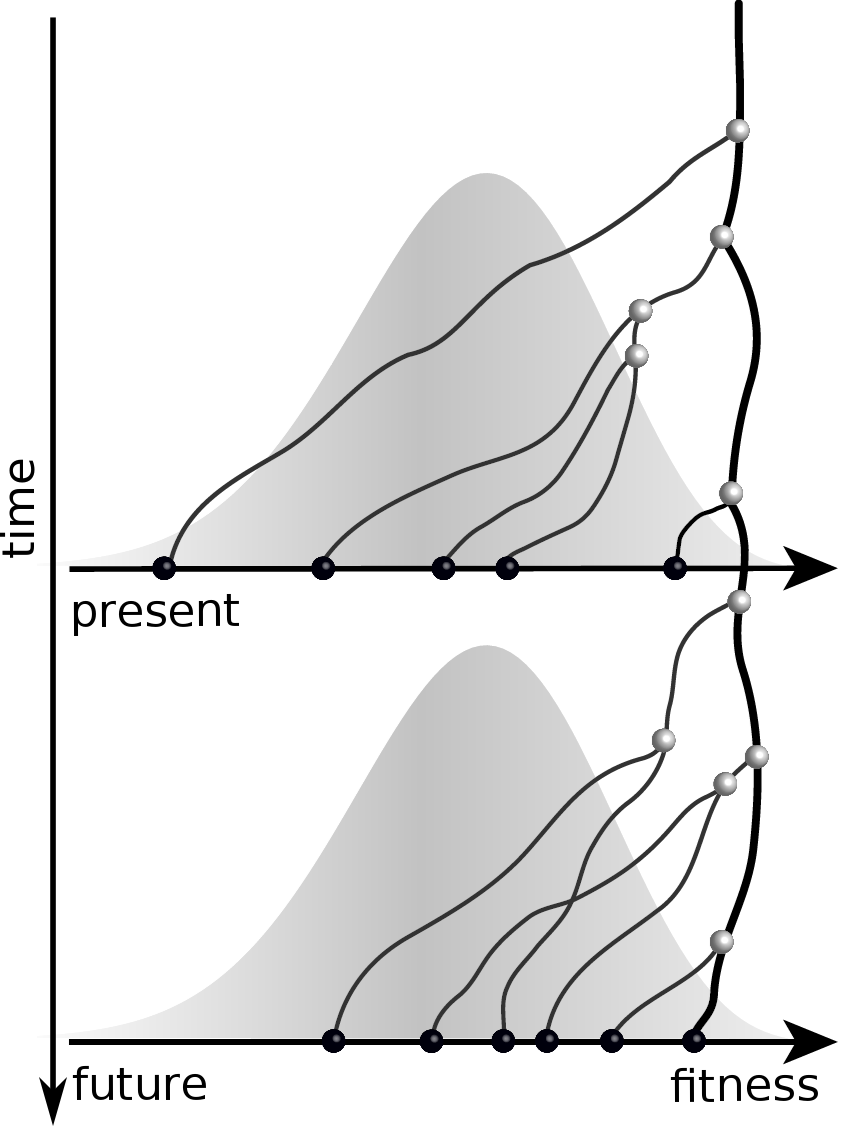
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
Validate on simulation data
- simulate evolution
- sample sequences
- reconstruct trees
- infer fitness
- predict ancestor of future
- compare to truth
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
Sequence alignment?
Reconstruction of phylogenetic trees
- There are super-exponentially many trees
- for $n$ taxa, there are $N = (2n-5)!! = (2n-5)*(2n-7)*\cdots*3*1$ trees
- There are efficient heuristics to reconstruct trees, e.g. Neighbor Joining

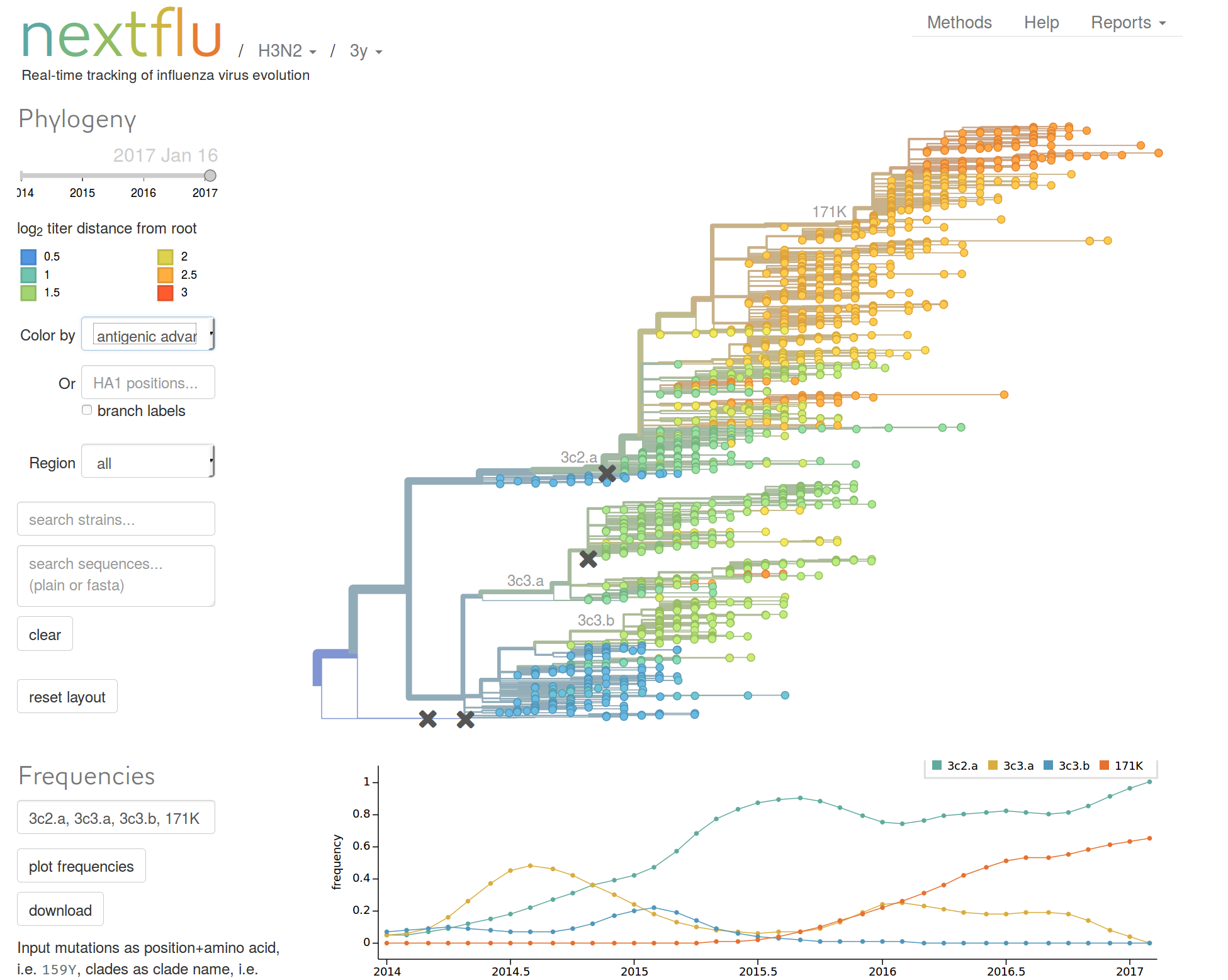
nextstrain.org
joint work with Trevor Bedford & his lab
code at github.com/nextstrain
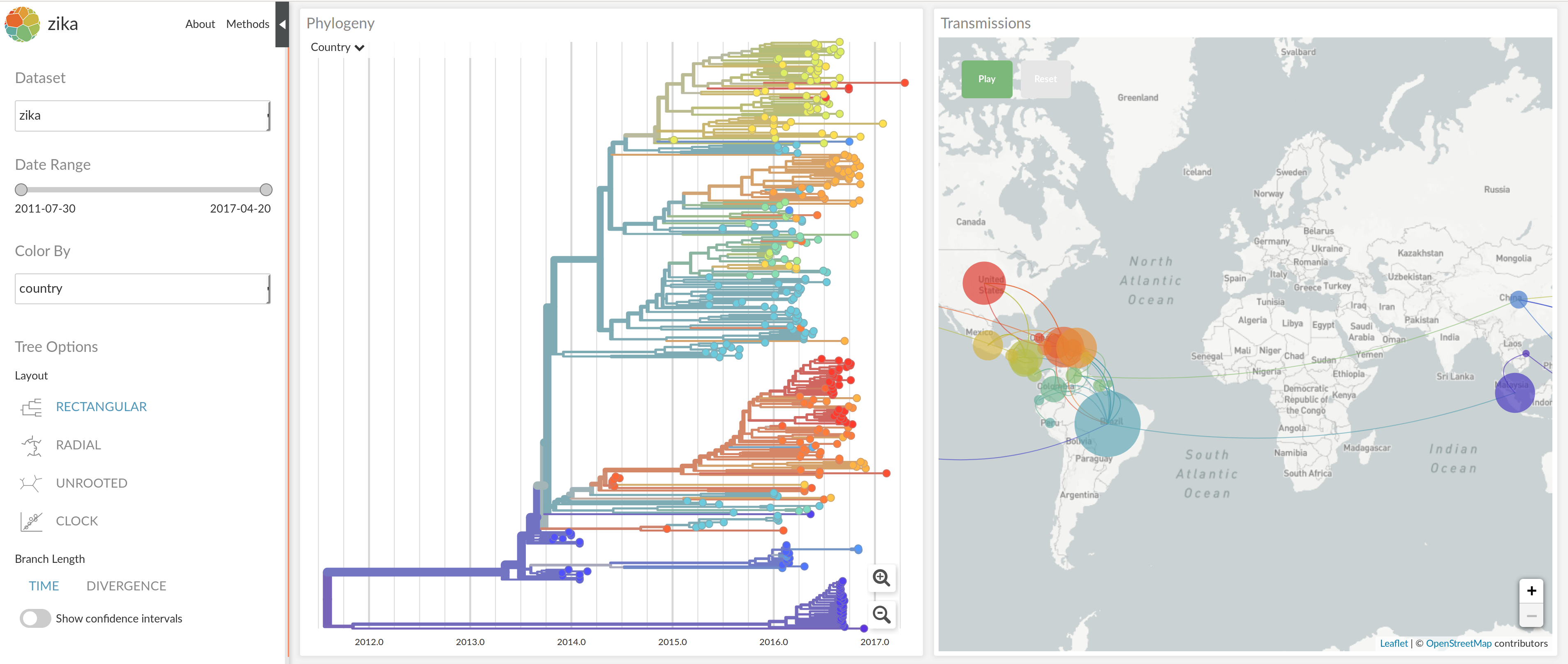
NextStrain architecture
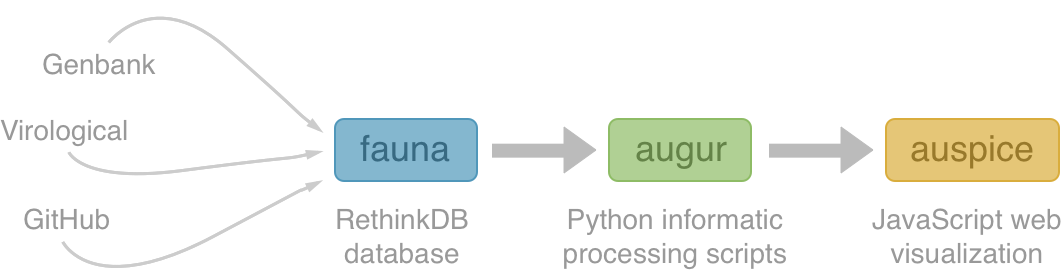
Using treetime to rapidly compute timetrees
TreeTime: maximum likelihood phylodynamic analysis
Phylogenetic trees record history:
- transmission
- divergence times
- population dynamics
- ancestral geographic distribution/migrations
Typical approach: Bayesian parameter estimation
- flexible
- probabilistic → confidence intervals etc
- but: computationally expensive
TreeTime by Pavel Sagulenko
- probabilistic treatment of divergence times
- dates trees with thousand sequences in a few minutes
- linear time complexity
- fixed tree topology
- github.com/neherlab/treetime
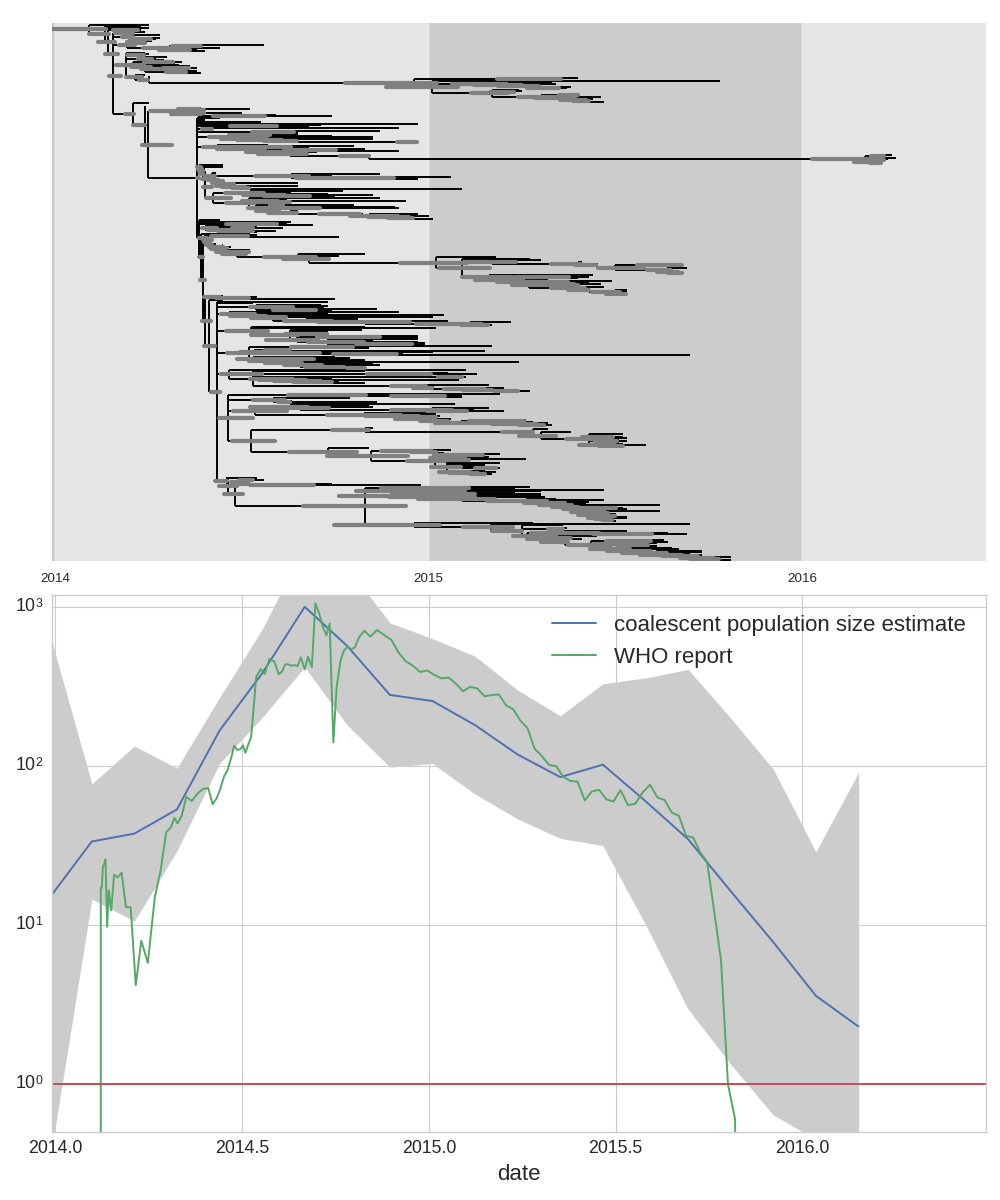
West African Ebola virus outbreak
TreeTime: nuts and bolts

Attach sequences and dates

Reconstruct ancestral sequences

Propagate temporal constraints via convolutions

Integrate up-stream and down-stream constraints
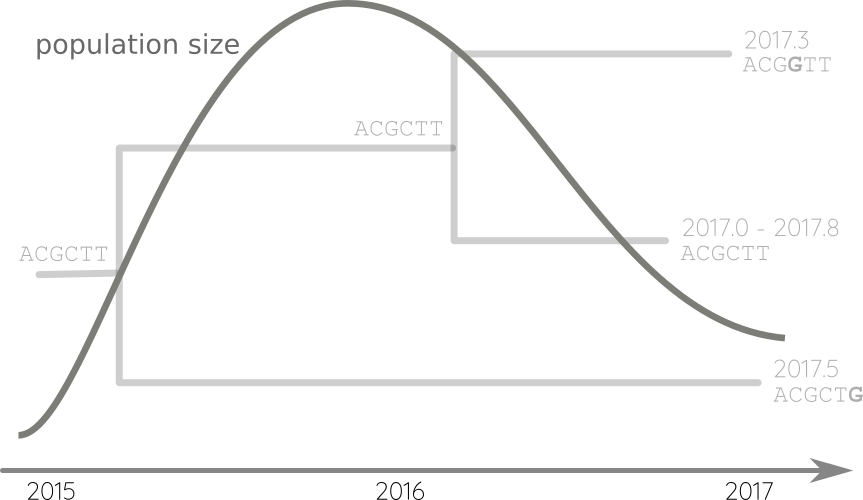
Fit phylodynamic model → iterate
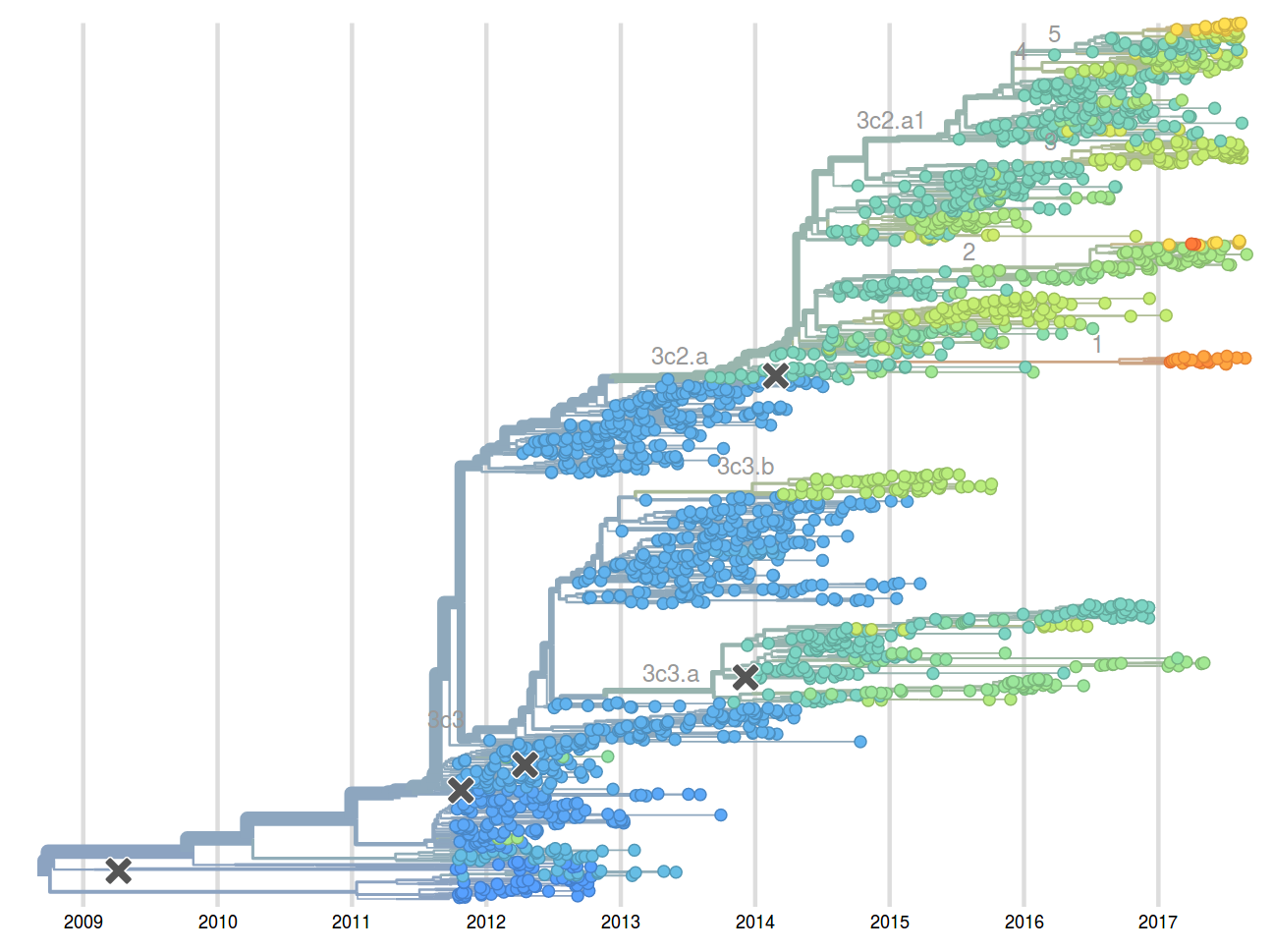
Molecular clock phylogenies of ~2000 A/H3N2 HA sequences -- a few minutes
What about bacteria?
- vertical and horizontal transmission
- genome rearrangements
- much larger genomes
- variation of divergence along the genome
- NGS genomes tend to be fragmented
- annotations of variable quality

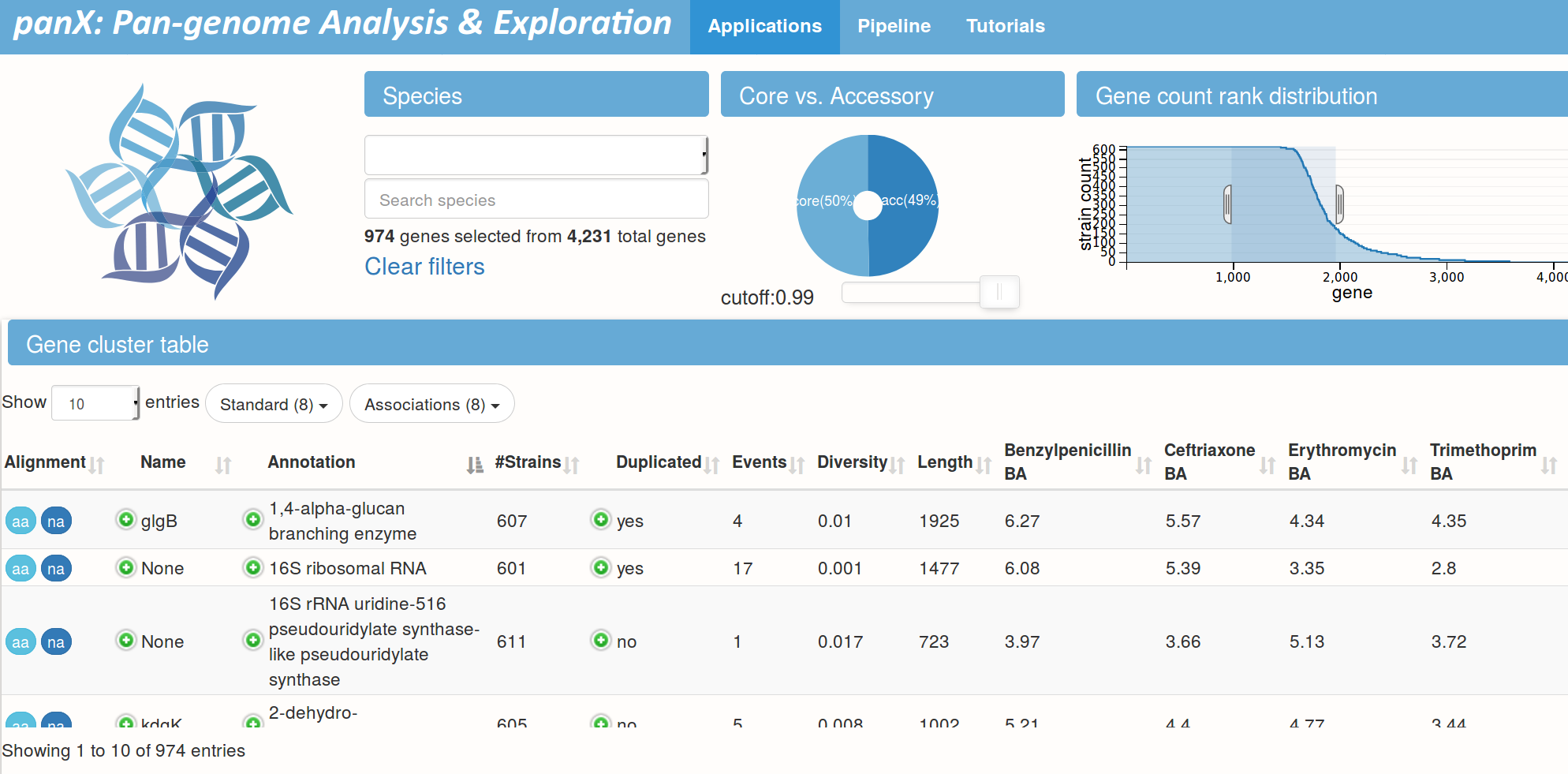
panX by Wei Ding
- pan-genome identification pipeline
- phylogenetic analysis of each orthologous cluster
- detect associations with phenotypes
- fast: analyze hundreds of genomes in a few hours
- github.com/neherlab/pan-genome-analysis
panX @ pangenome.de
S. pneumoniae data set by Croucher et al.
Pan-genome statistics and filters
Species trees and gene trees
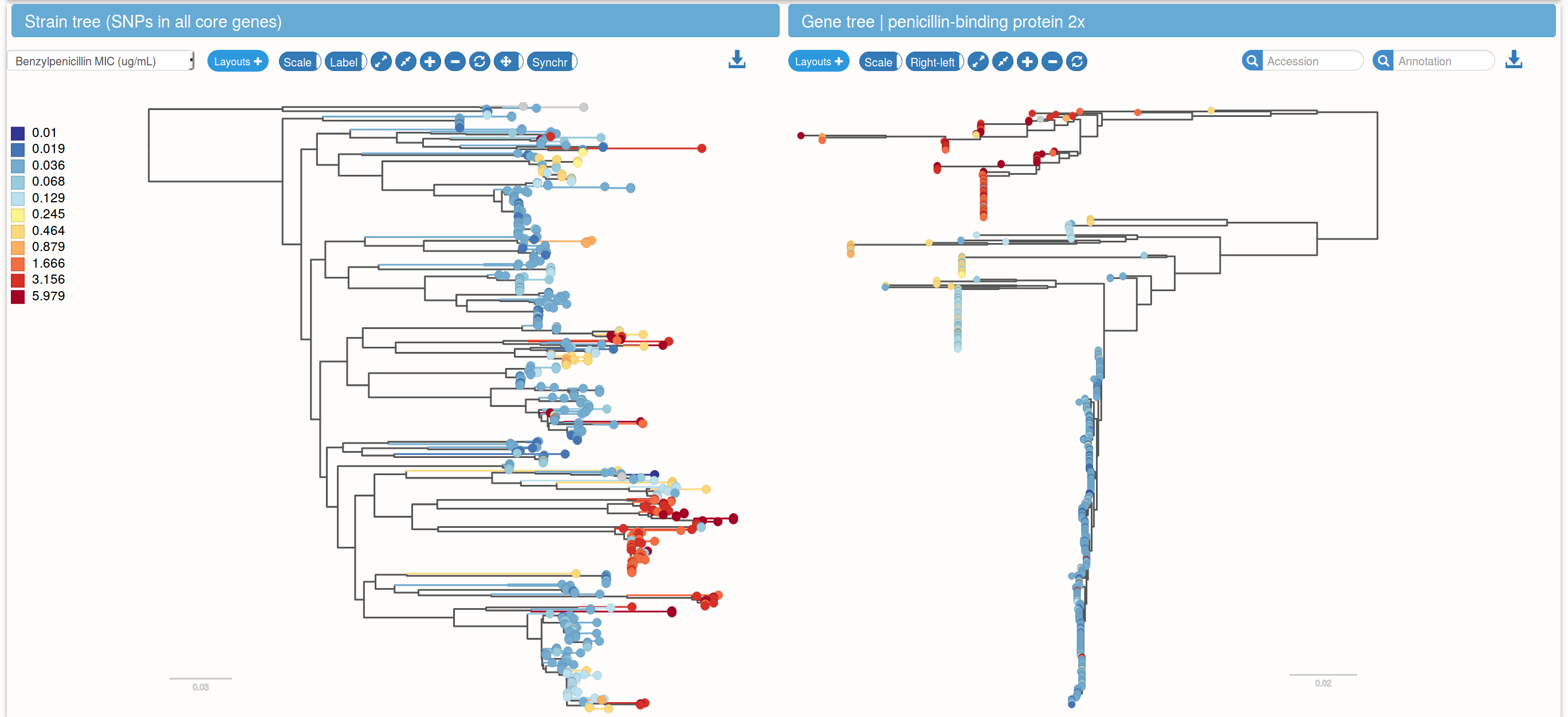
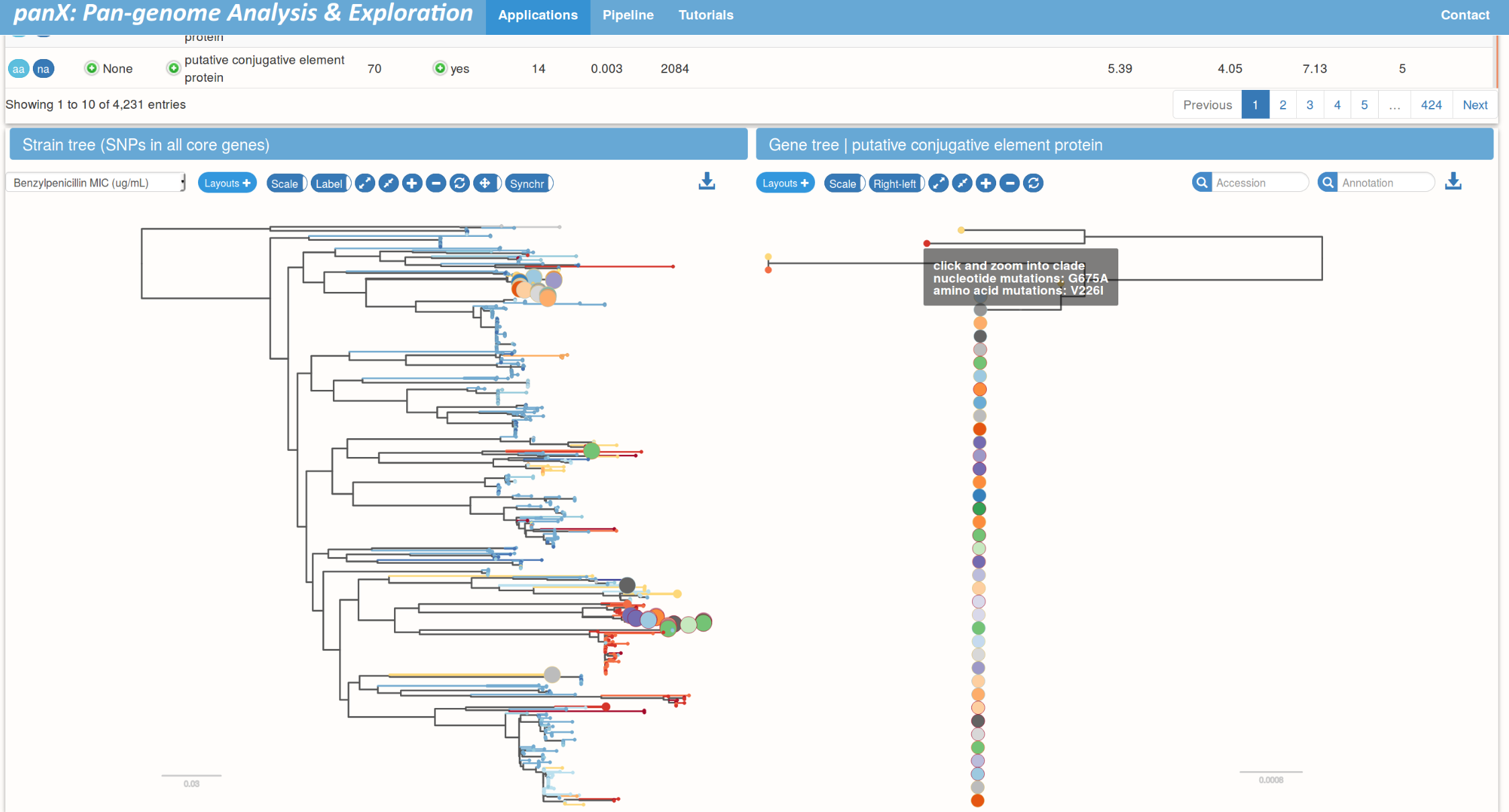
Links between species trees and gene trees
Summary
- Data set are growing rapidly
→ tools for interpretation and exploration are crucial - Breadth and depth
→ provide an overview, integrate, and go deep - Actionable outputs require (near)real-time analysis
→ fast analysis pipelines are essential - We are just scratching the surface...
Interested in HIV NGS: come find me!
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
- All the NICs and WHO CCs that provide influenza sequence data
nextstrain.org



- Trevor Bedford
- Colin Megill
- Sidney Bell
- James Hadfield
- All the scientist that share virus sequence data

TreeTime & panX

TreeTime: Pavel Sagulenko
github.com/neherlab/treetimewebserver at treetime.ch
manuscript on bioRxiv
panX: Wei Ding
github.com/neherlab/pan-genome-analysislive site at pangenome.de
manuscript on bioRxiv