Learning about evolution from pathogen sequence data
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201805_InHouse.html
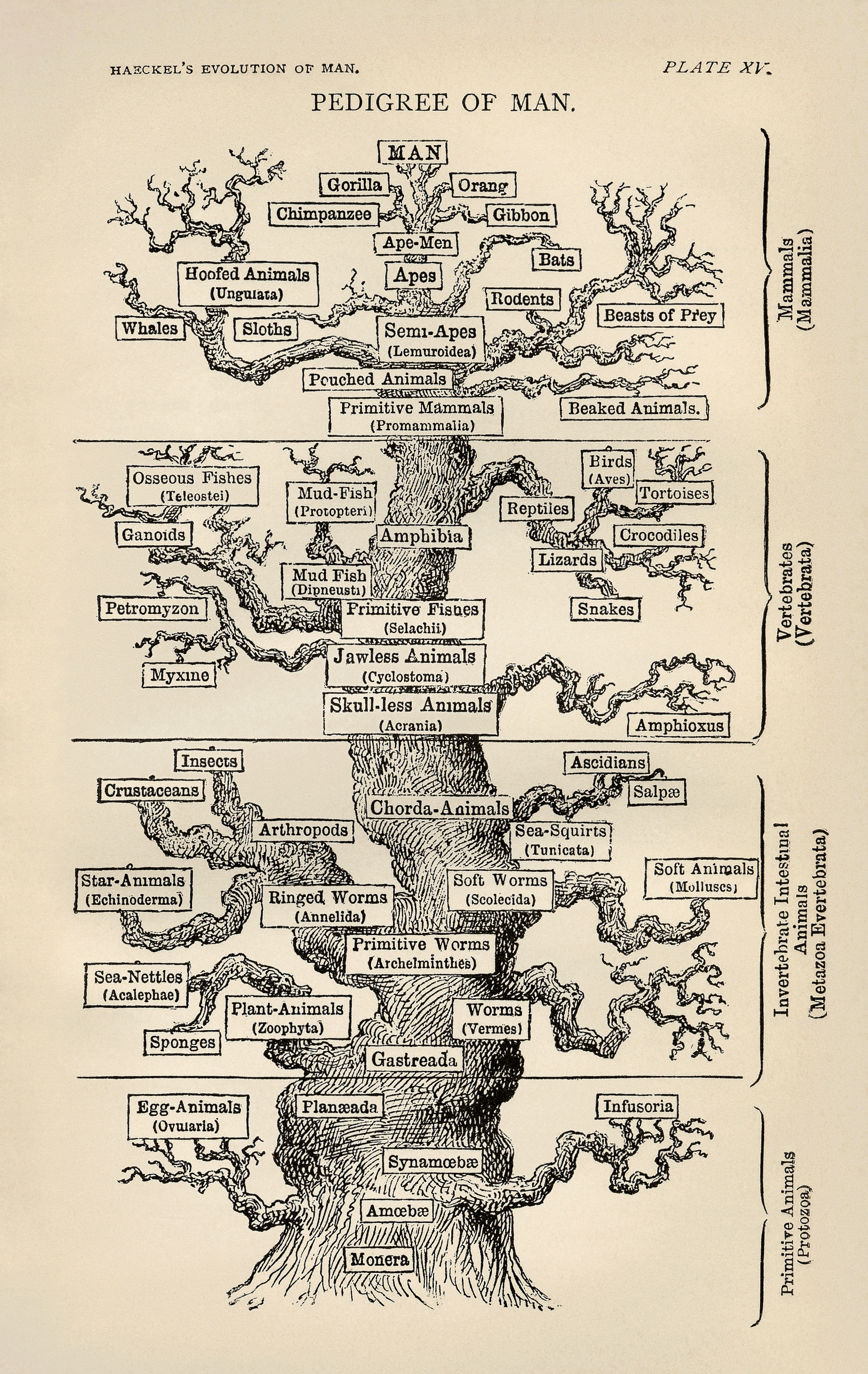 Ernst Haeckel, 1879
Ernst Haeckel, 1879
A quantitative understanding of evolution?
- How repeatable is evolution?
- How gradual is evolution?
- How predictable is evolution?
- What are the relevant parameters?
(population size, mutation rates etc) - How do the dynamics depend on parameters?
Challenges
- Most data are static snap shots.
- Genotype-phenotype map is largely unknown.
- Ecology and environments are complex and variable.
Experimental evolution -- Lenski experiment
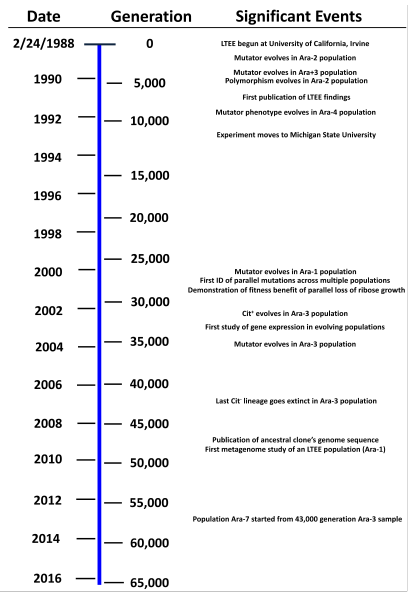

 Experiment started 1927, one drop every 10 years. wikipedia.org
Experiment started 1927, one drop every 10 years. wikipedia.org

Influenza A/H3N2
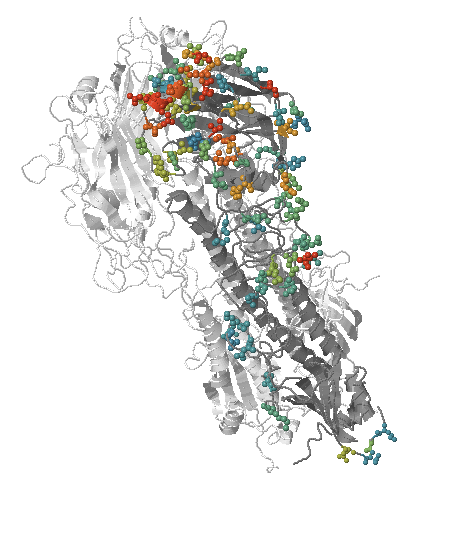
- Influenza viruses evolve to avoid human immunity
- Vaccines need frequent updates
Rapidly evolving RNA viruses -- HIV

Evolution of HIV
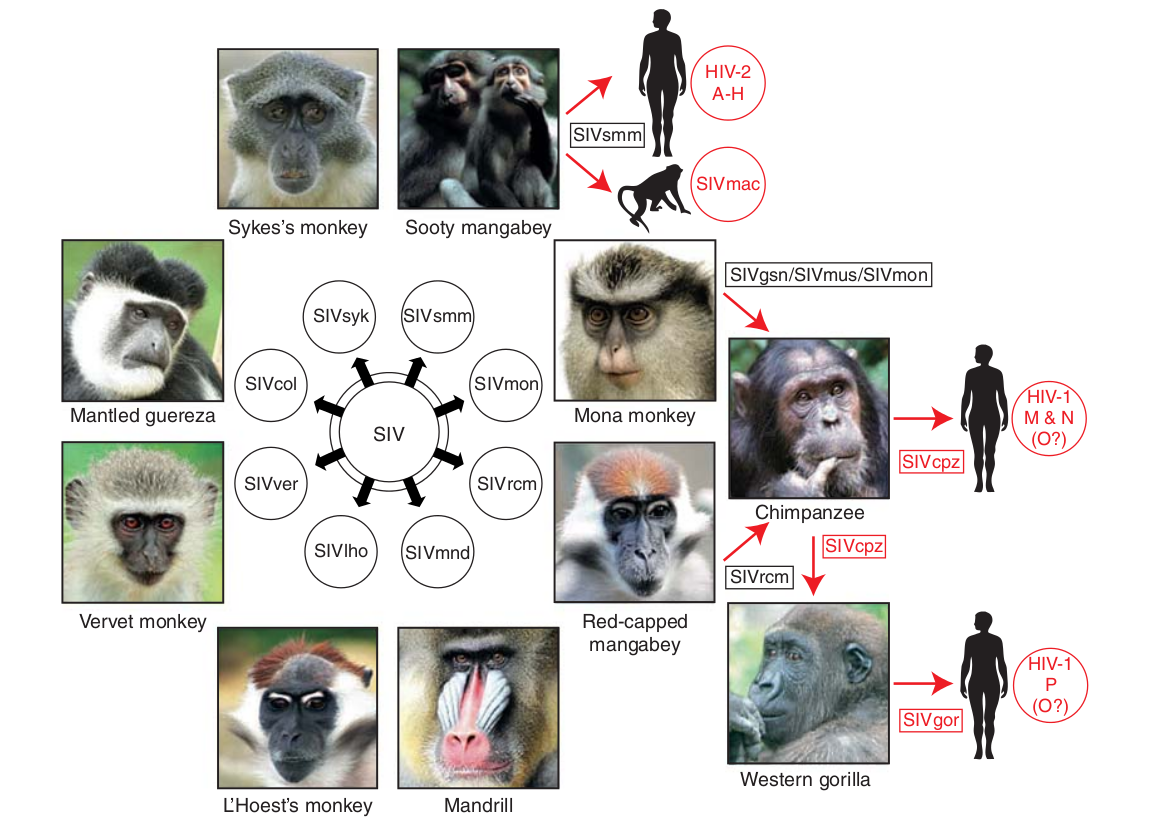
- Chimp → human transmission around 1900 gave rise to HIV-1 group M
- ~100 million infected people since
- subtypes differ at 10-20% of their genome
- HIV-1 evolves ~0.1% per year
HIV infection
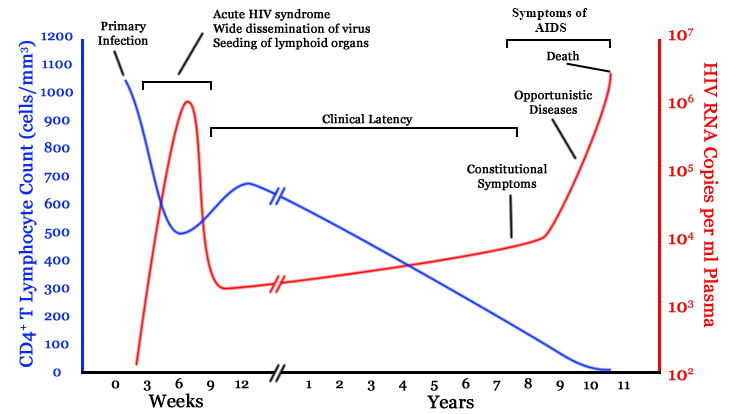

- $10^8$ cells are infected every day
- the virus repeatedly escapes immune recognition
- integrates into T-cells as
latent provirus
{kind=link}
HIV acknowledgments







- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller
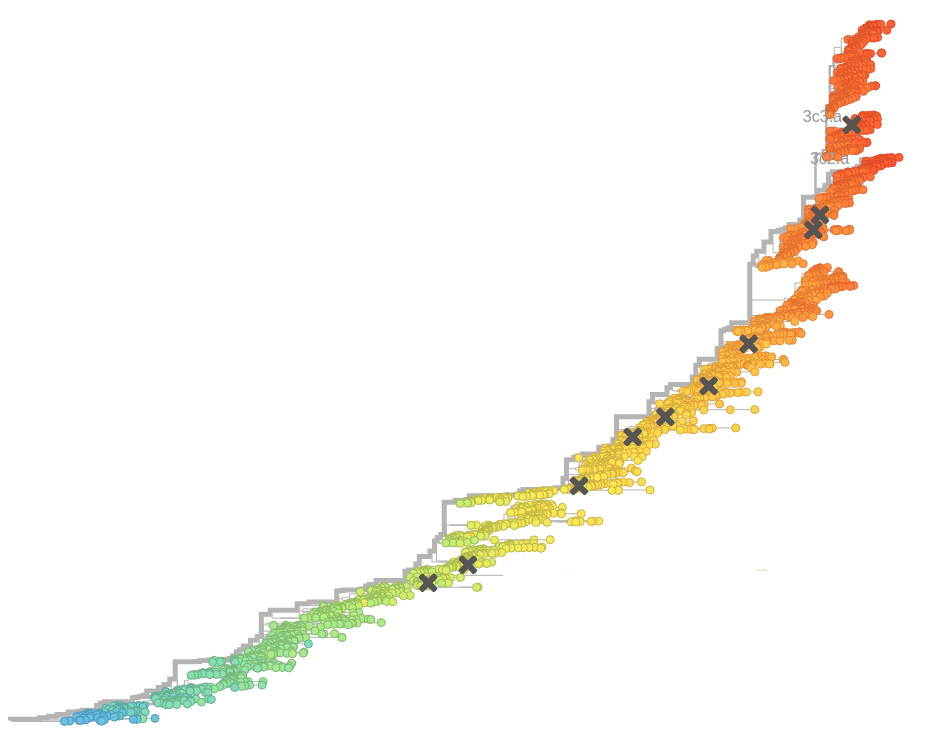
HIV-1 evolution within one individual
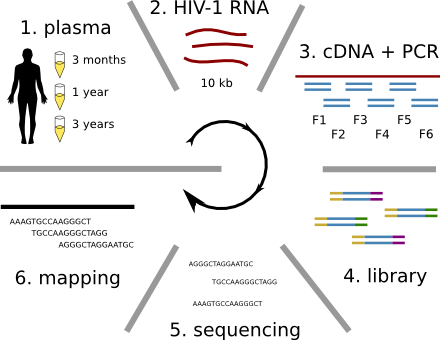

HIV-1 sequencing before and after therapy
 Zanini et al, eLife, 2015;
Brodin et al, eLife, 2016.
Collaboration with the group of Jan Albert
Zanini et al, eLife, 2015;
Brodin et al, eLife, 2016.
Collaboration with the group of Jan Albert
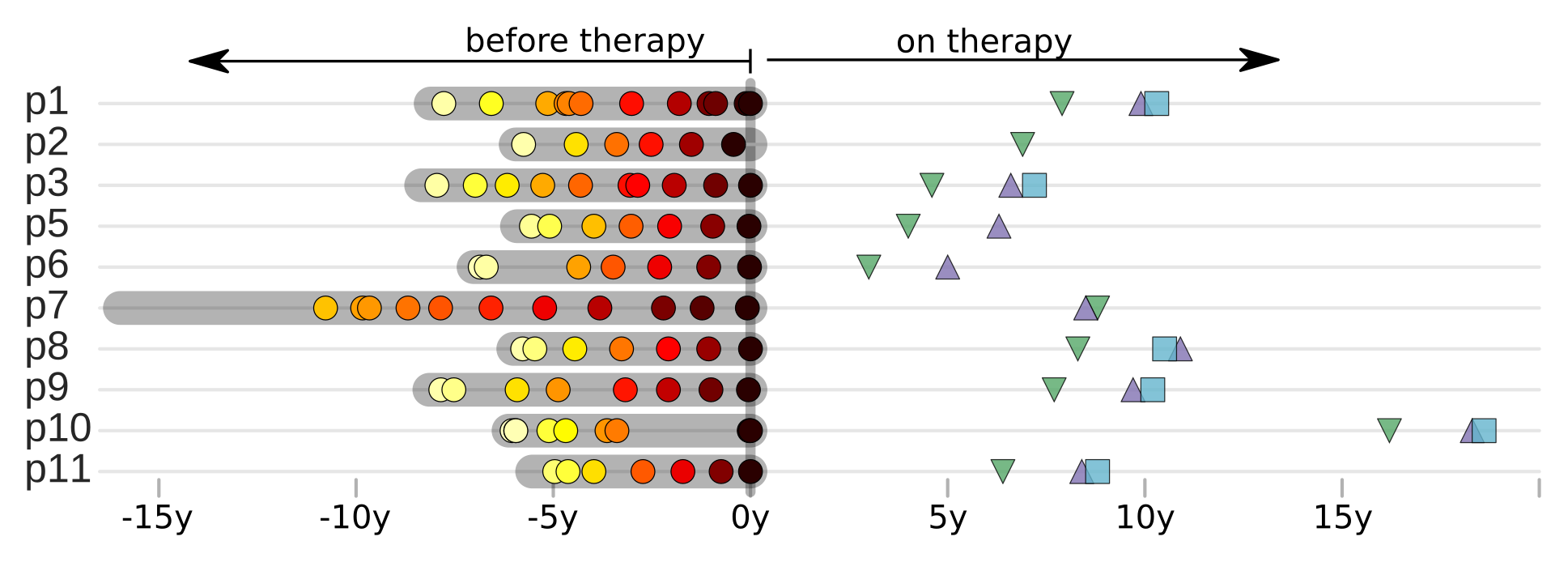
Estimating the date of infection from diversity data
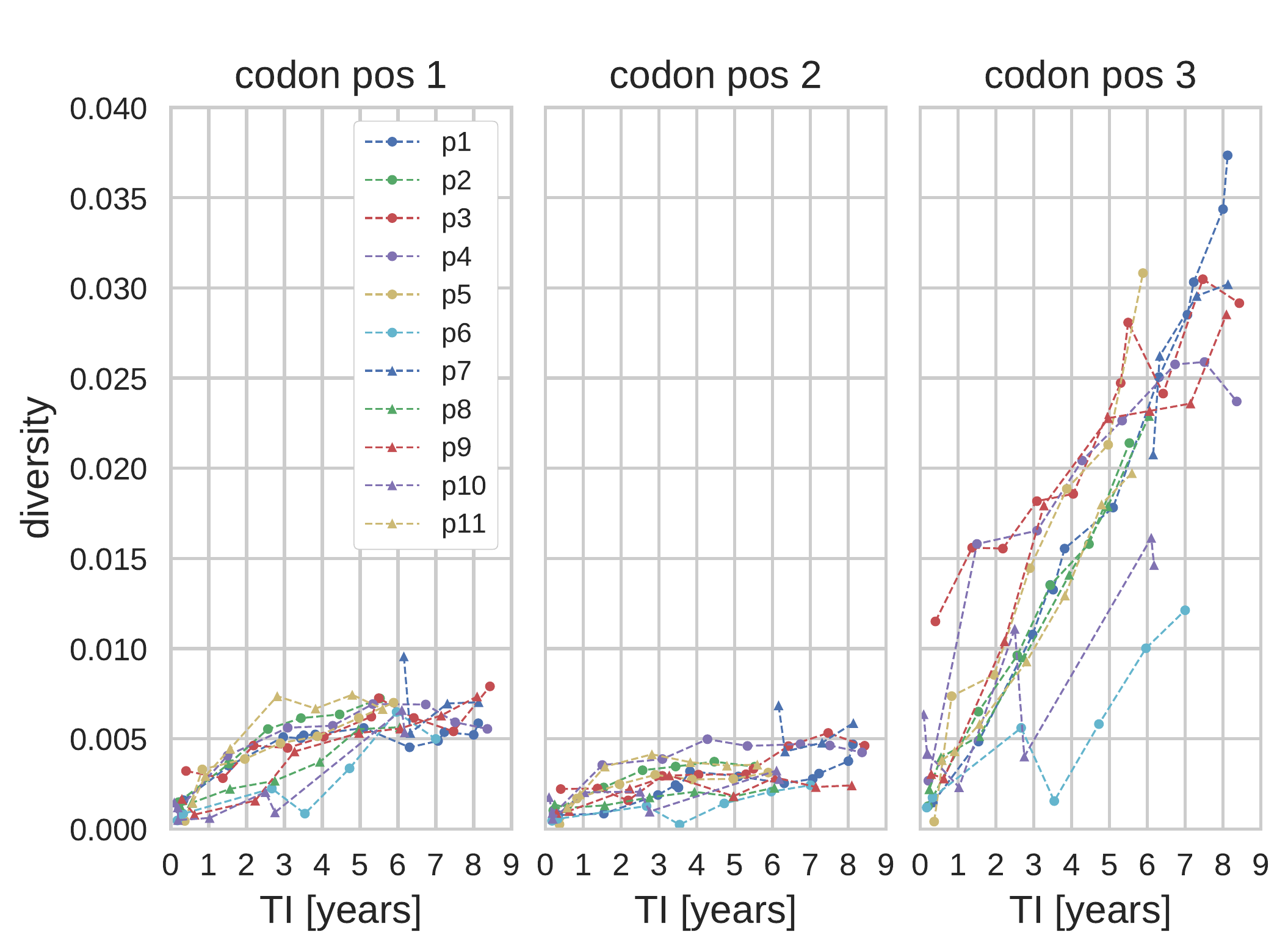
- diversity at 3rd positions increases almost linearly in time
- can be used to predict date of infection
- critical to estimate incidence
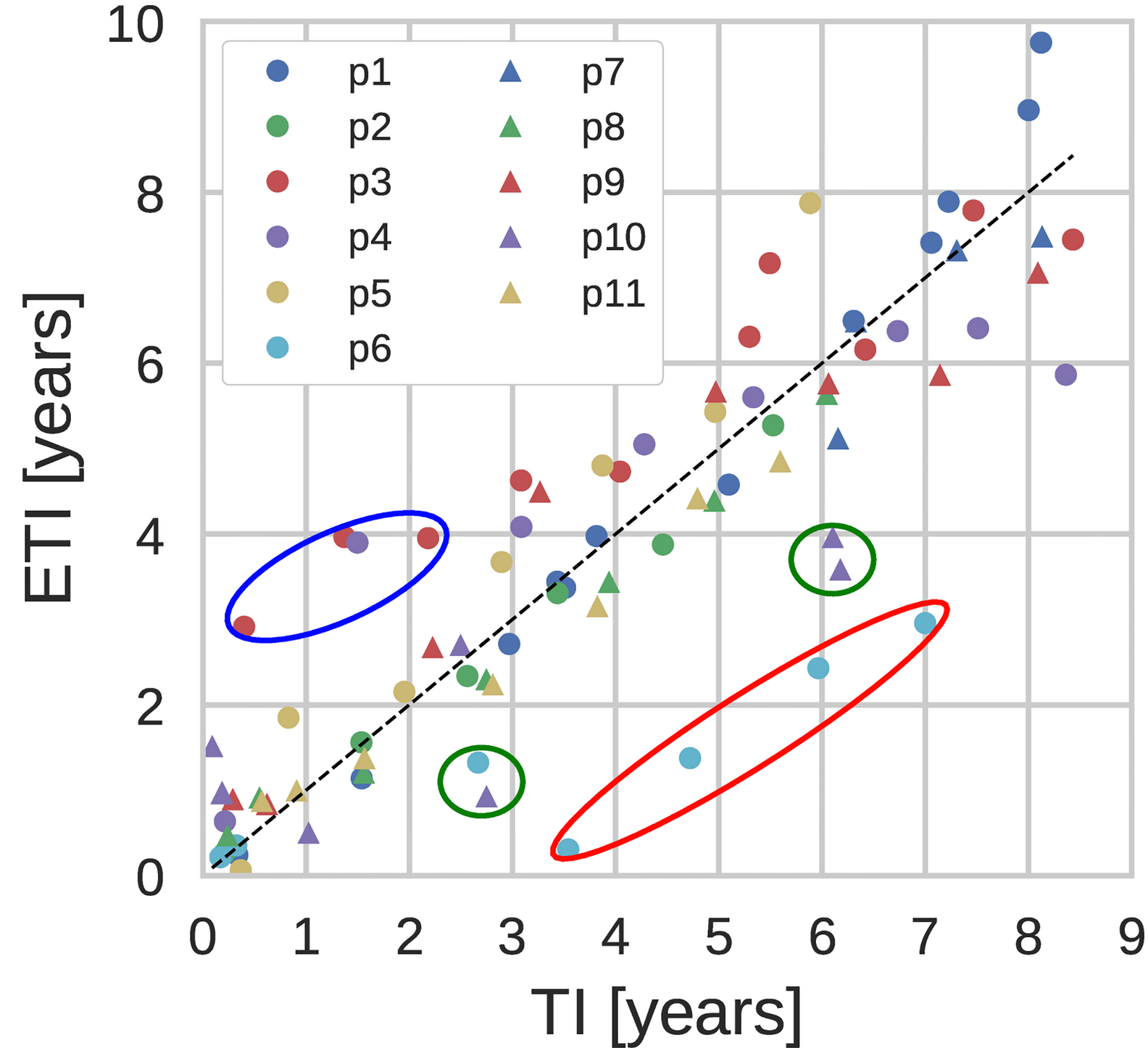
- Multiple founder viruses cause over estimation
- Degraded samples cause under estimation
Inference of fitness costs
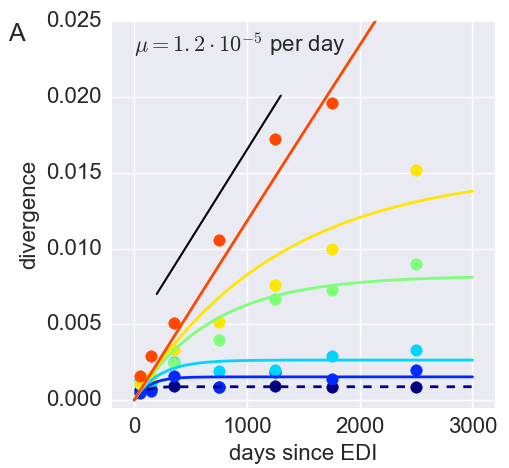
- mutation away from preferred state with rate $\mu$
- selection against non-preferred state with strength $s$
- variant frequency dynamics: $\frac{d x}{dt} = \mu -s x $
- equilibrium frequency: $\bar{x} = \mu/s $
- fitness cost: $s = \mu/\bar{x}$
Fitness landscape of HIV-1
Zanini et al, Virus Evolution, 2017Selection on RNA structures and regulatory sites
- red: mutations that don't change protein sequence
- blue: all mutations
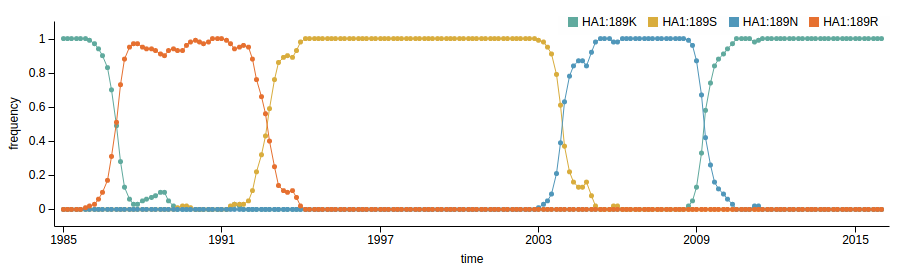
Influenza A/H3N2 virus evolution
Joint work with....



- Boris Shraiman
- Colin Russell
- Trevor Bedford
nextflu.org
joint work with Trevor Bedford & his lab
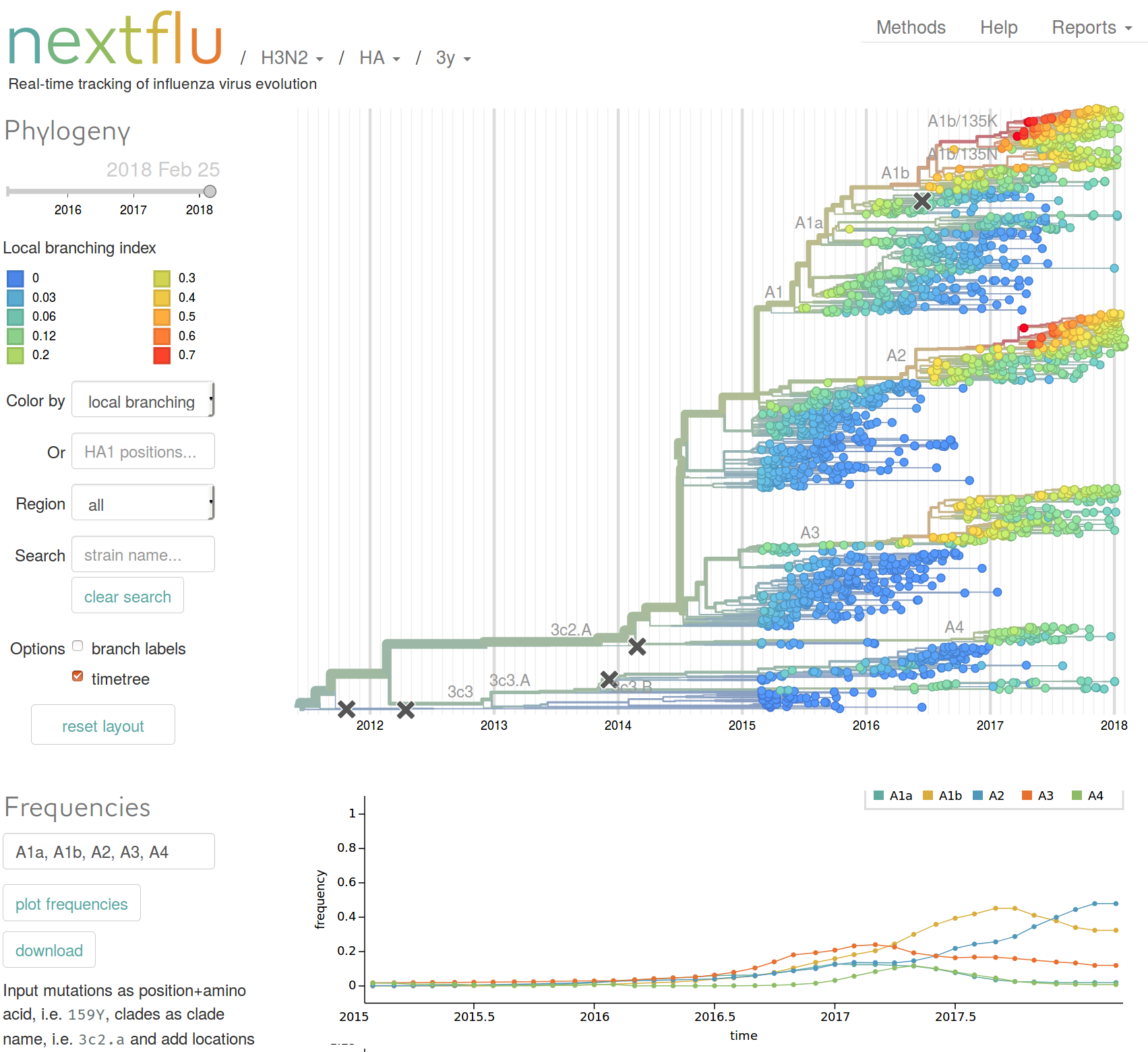
Prediction of the dominating H3N2 influenza strain
RN, Russell, Shraiman, eLife, 2014nextstrain.org






- Trevor Bedford
- Colin Megill
- Pavel Sagulenko
- Sidney Bell
- James Hadfield
- Wei Ding
- Emma Hodcroft

nextstrain.org
joint work with Trevor Bedford & his lab
nextstrain.org
- integrate data from many different sources
- analyze those data in near real time
- disseminate results in an intuitive yet informative way
- provide actionable insights
What about more complicated things than viruses?
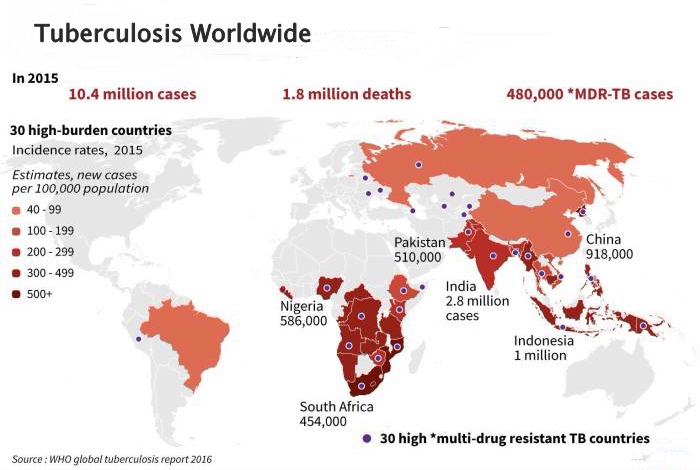
nextTB: real-time molecular epidemiology of TB
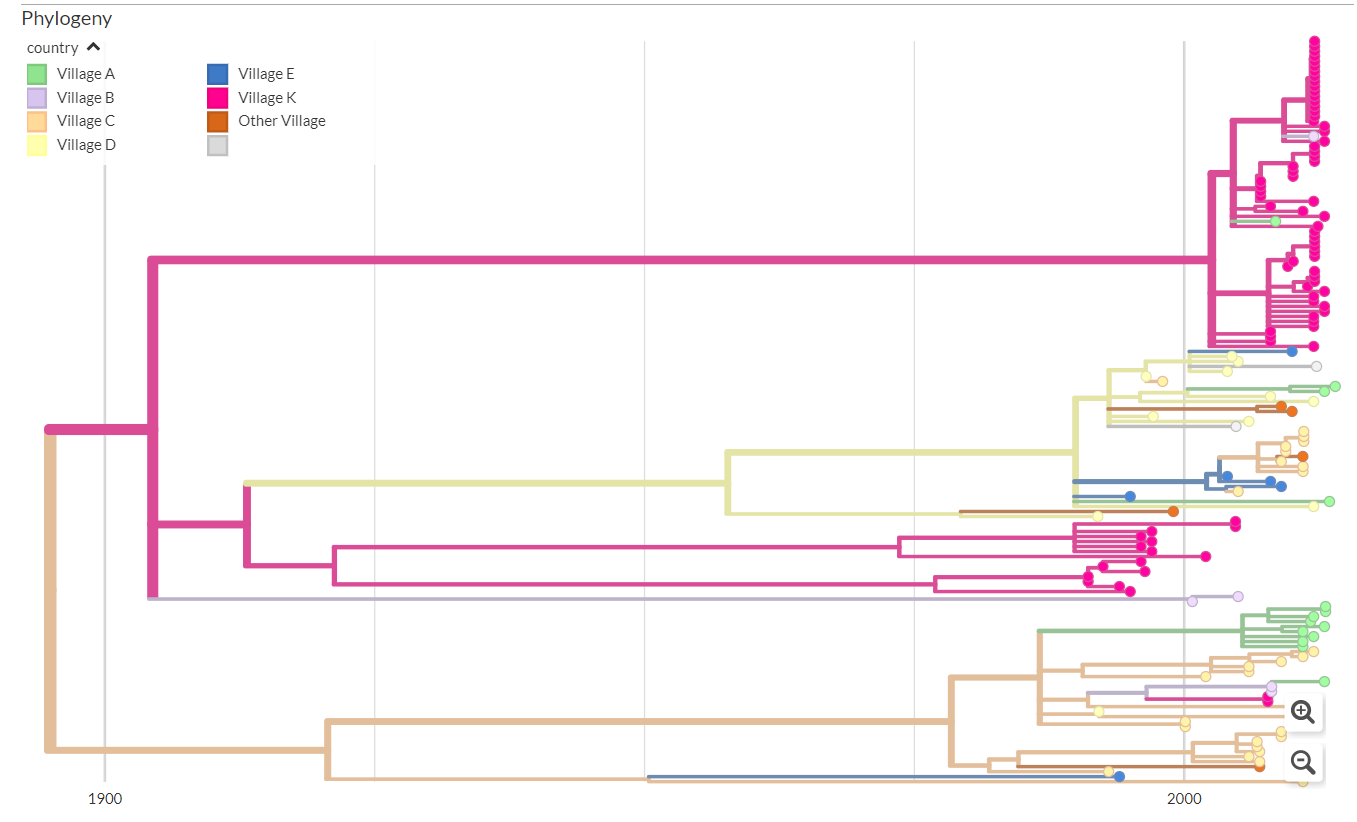
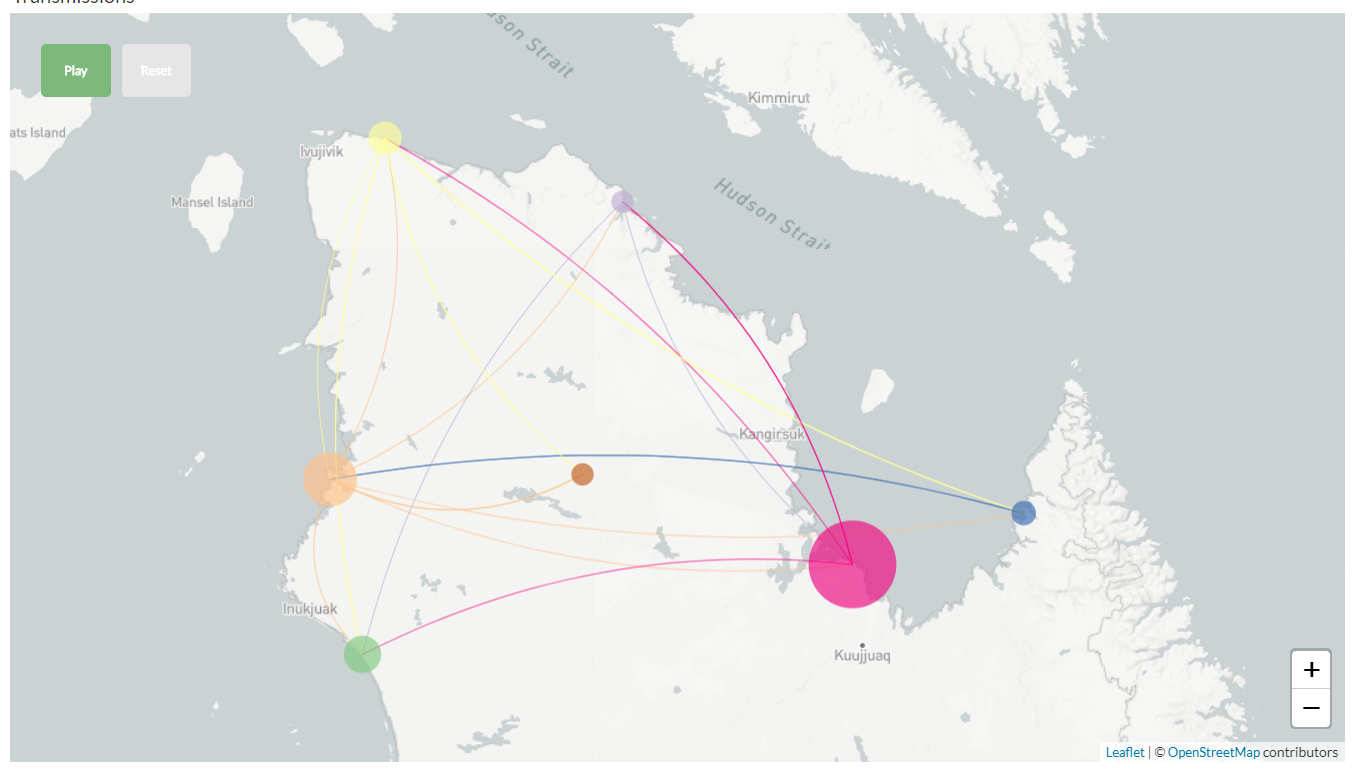
Collaboration with Sebastian Gagneux and colleages at the STPH
- 10s of thousand MTB genomes have been sequenced
- Elucidate transmission routes at the local and global level
- Integrate with drug resistance data
Pan-genomes of bacteria
- much larger genomes
- vertical and horizontal transmission
- gene gain and loss
- genome rearrangements
- variation of divergence along the genome
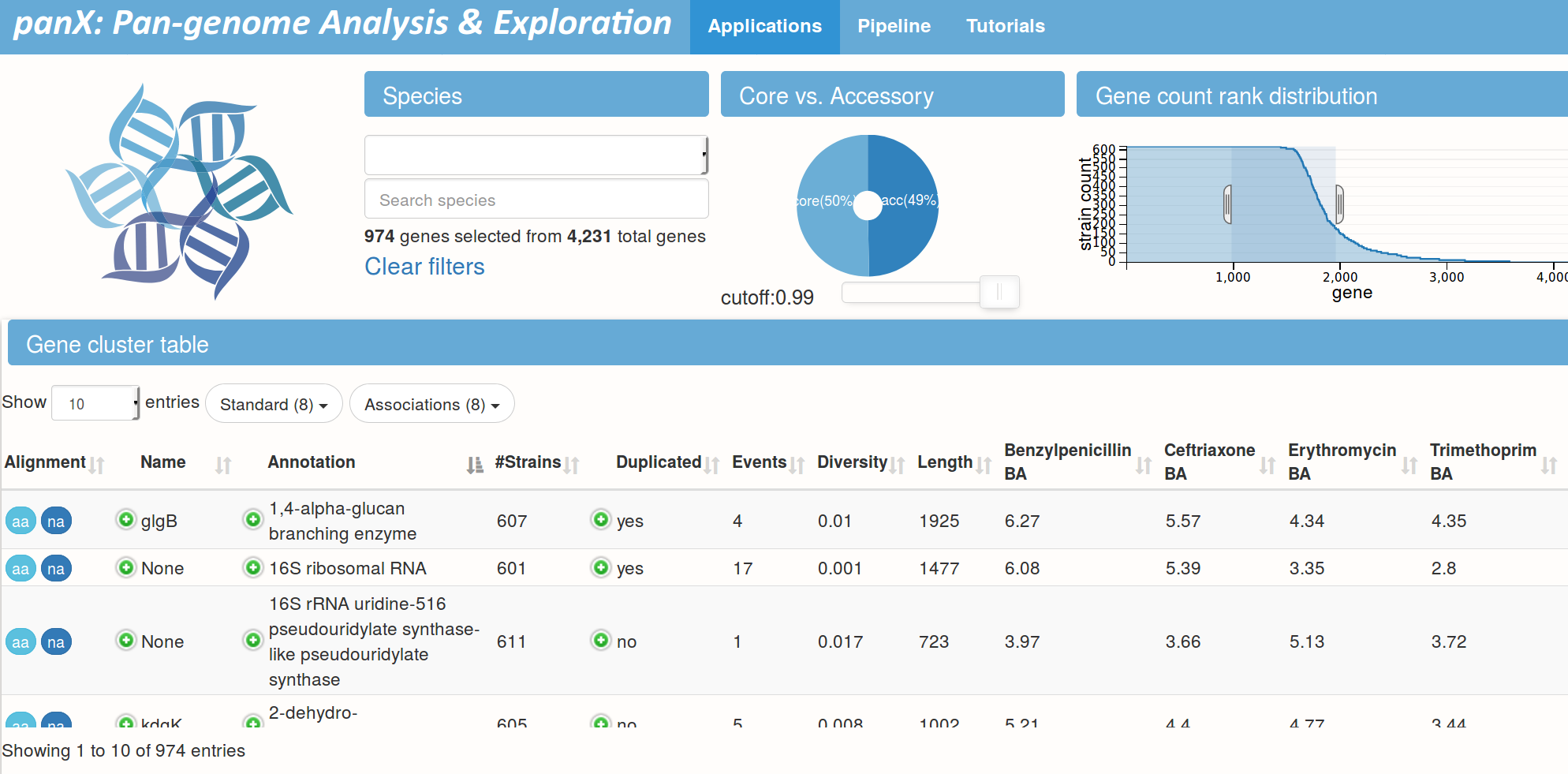
panX by Wei Ding
- pan-genome identification pipeline
- phylogenetic analysis of each orthologous cluster
- detect associations with phenotypes
- fast: analyze hundreds of genomes in a few hours
- pangenome.ch
- github.com/neherlab/pan-genome-analysis
Pan-genome statistics and filters
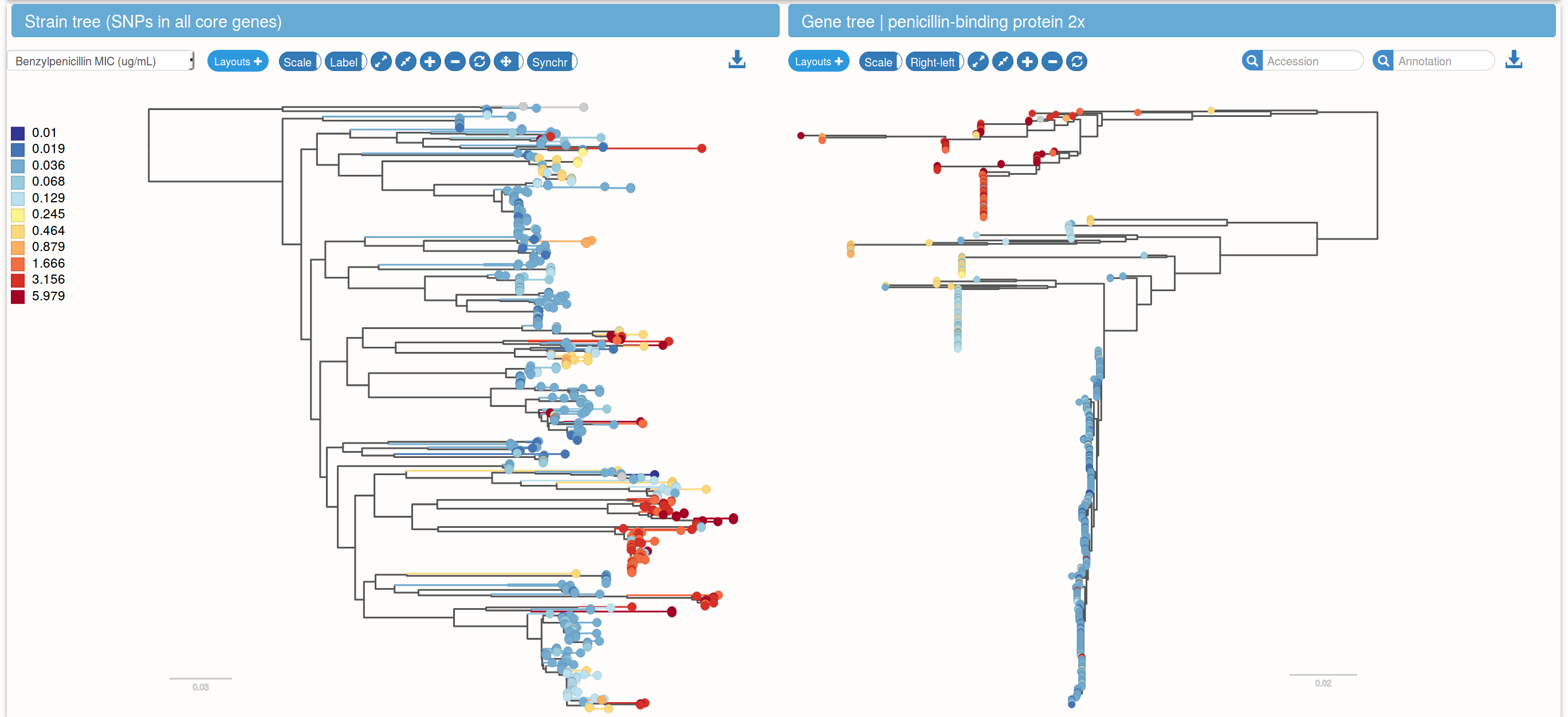
Species trees and gene trees
Genome assembly with short reads

- 10s of millions of short reads (<500bp)
- Too short to bridge repetitive elements
- → assemblies are fragmented into 100s of "contigs"
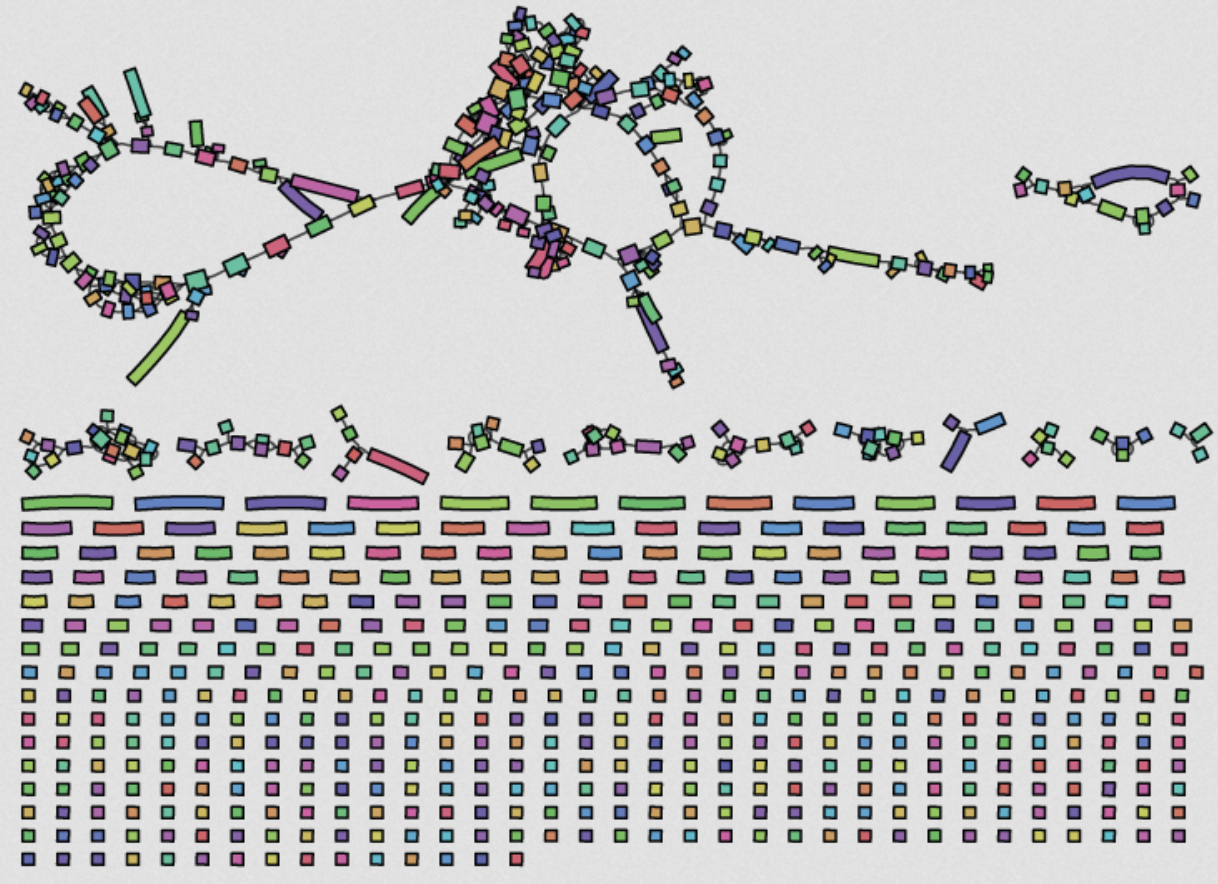
(really terrible example)
Long-read sequencing



Acknowledgements
Group
- Fabio Zanini
- Pavel Sagulenko
- Vadim Puller
- Wei Ding
- Sanda Dejanic
- Emma Hodcroft
- Nicholas Noll
- Eric Ulrich
Karolinska Institute
- Jan Albert
- Lina Thebo
- Johanna Brodin
FHCRC
- Trevor Bedford
- James Hadfield
- Sidney Bell
- Colin Megill
Swiss TPH
- Sebastian Gagneux
- Chloé Loiseau
- Fabrizio Menardo
USB
- Adrian Egli
- Daniela Lang
BZ
- Leo Faletti
- sciCore
- workshops
- Boris Shraiman (UCSB)
- Colin Russell (AMC)
- Oskar Hallatschek (UCB)