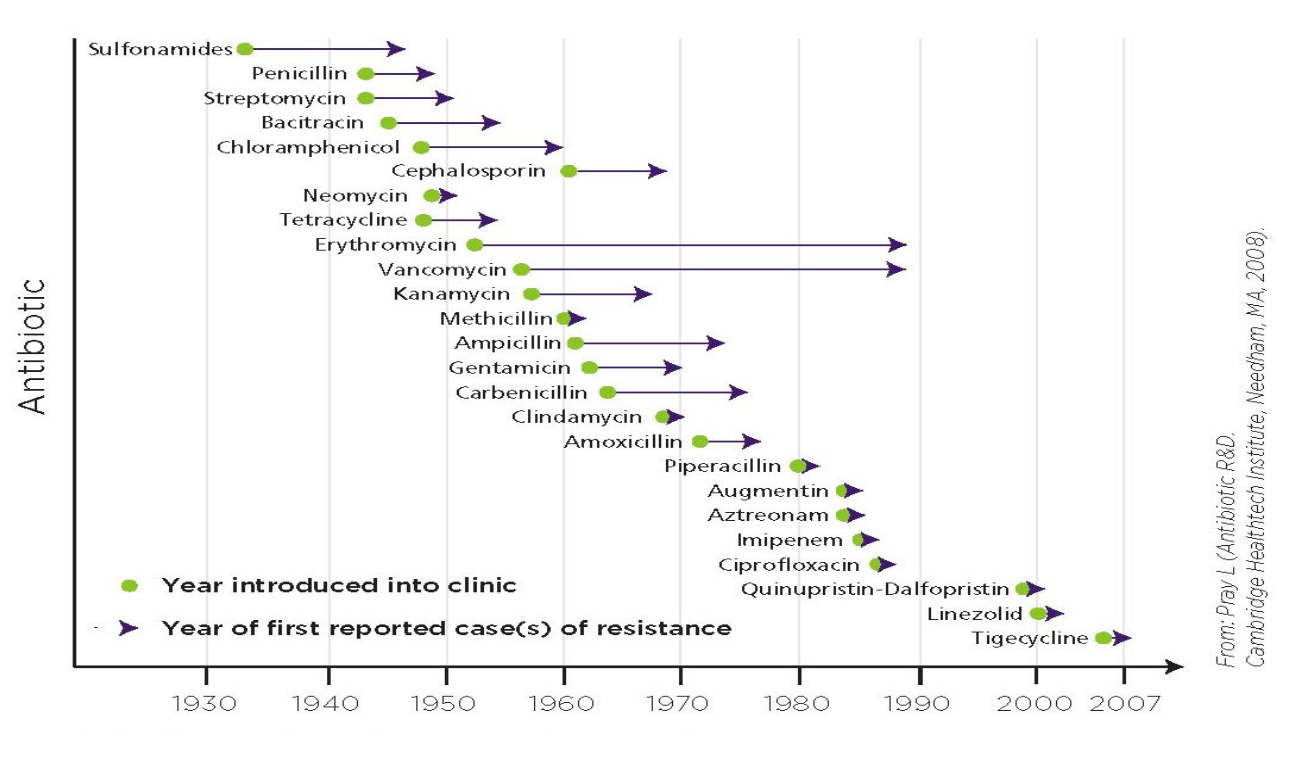
Microbial evolution
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201806_Paros.html
Human seasonal influenza viruses
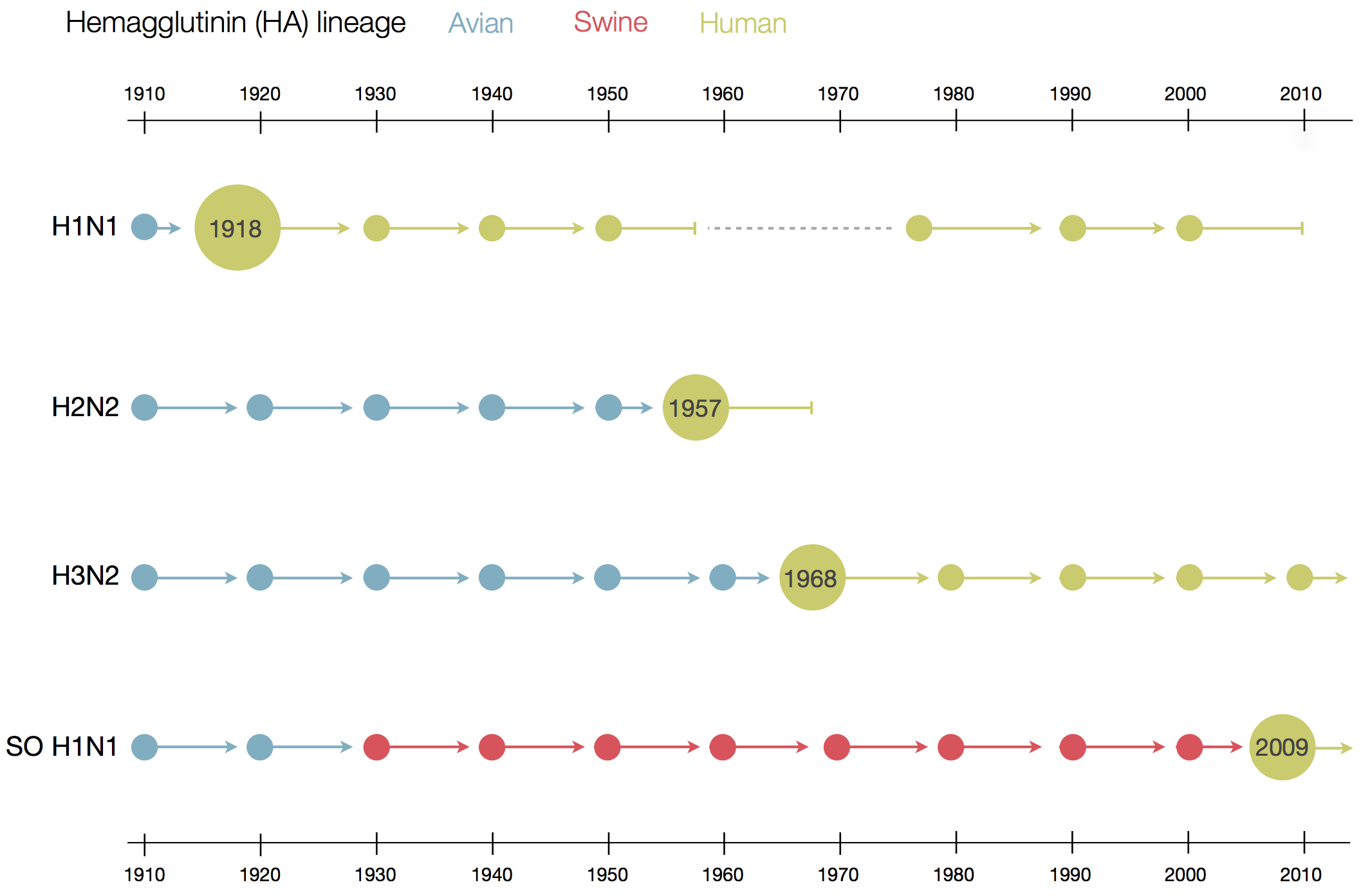
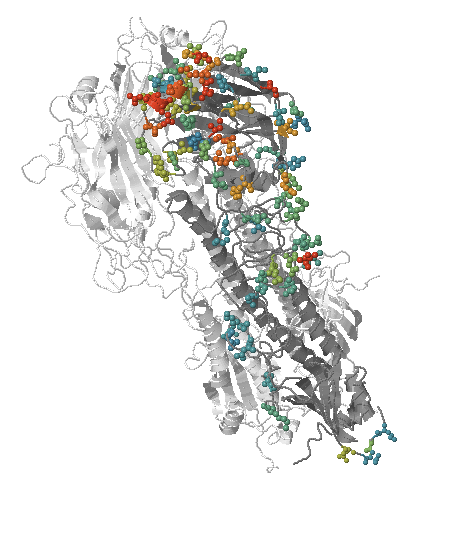
- Influenza viruses evolve to avoid human immunity
- Vaccines need frequent updates
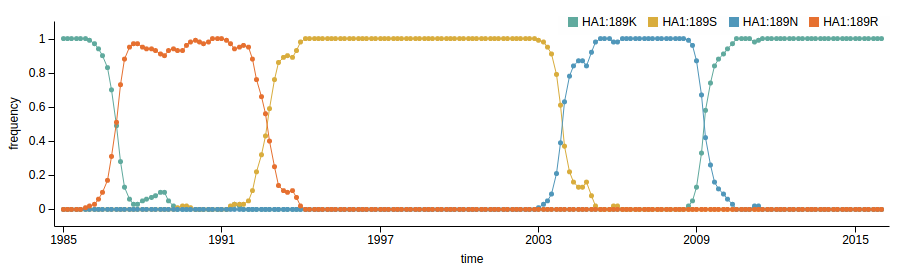
Model of rapidly adapting virus populations
Typical tree
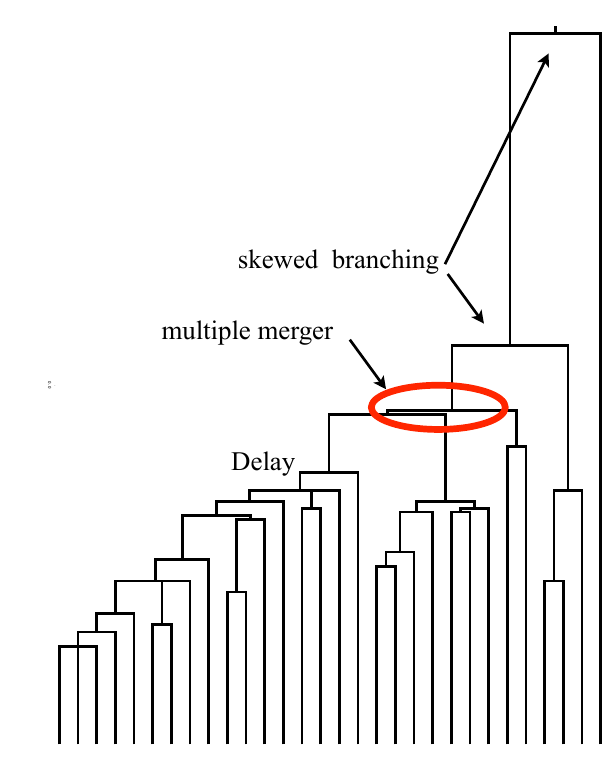
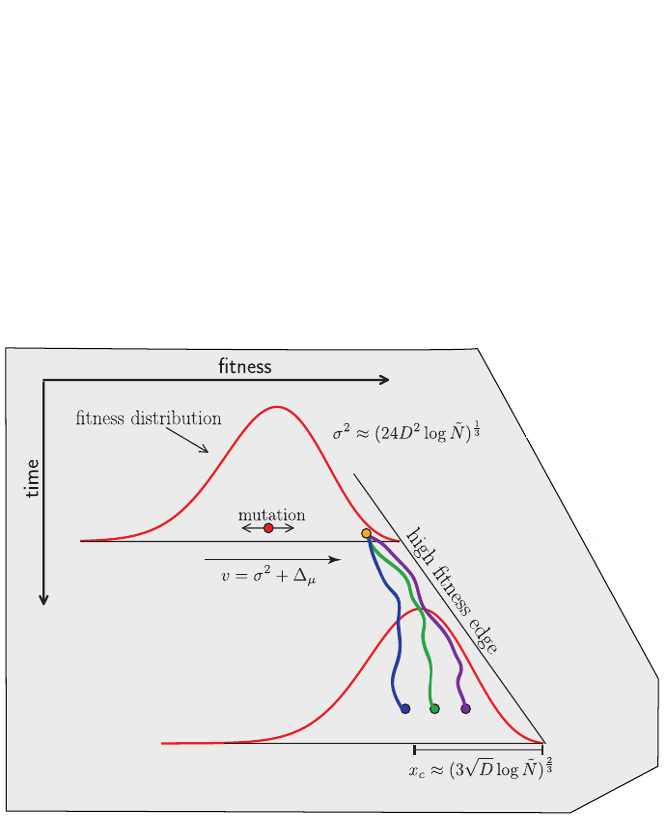
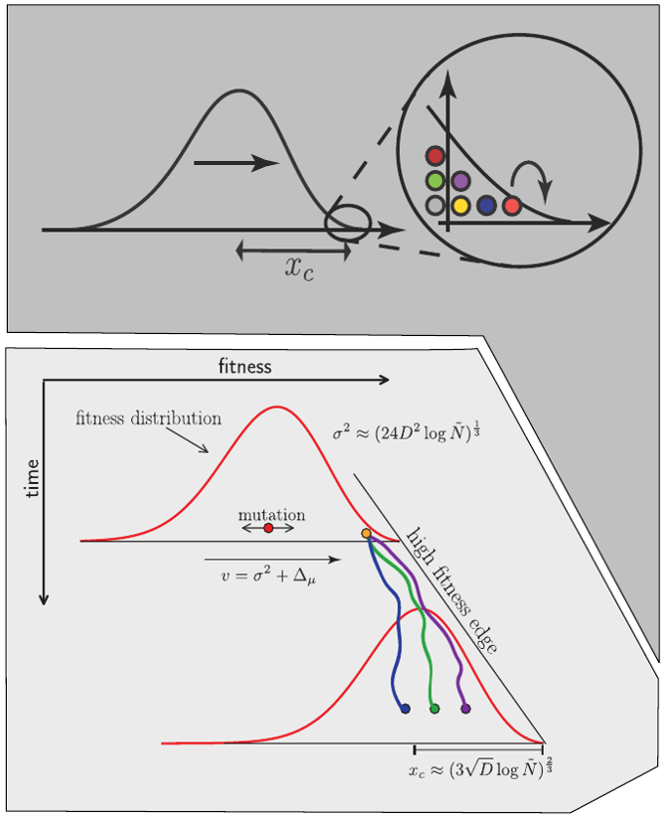
Bolthausen-Sznitman Coalescent
Traveling waves and the Bolthausen-Snitman coalescent
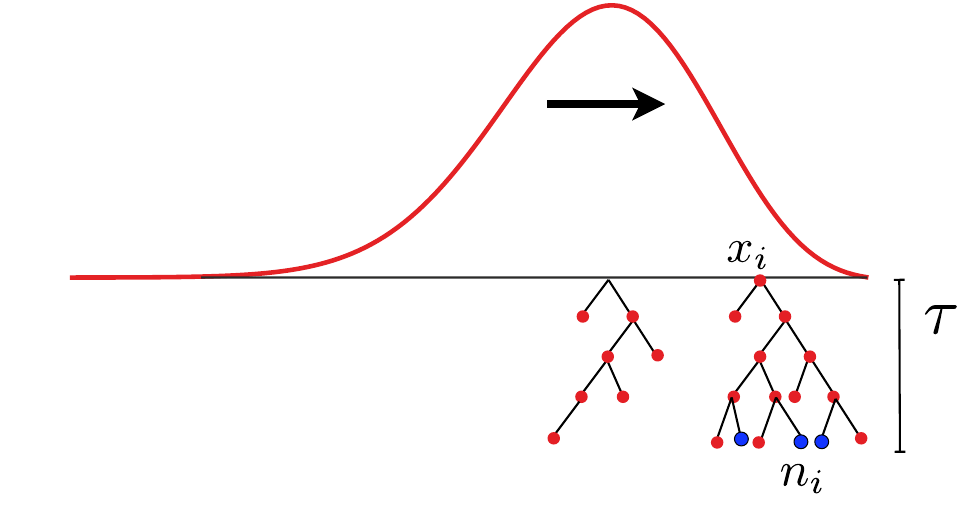
- Branching process approximation: $P(n_i, t|x_i)$
-
Does a sample (blue dots) have a common ancestor $\tau$ generations ago?
$\quad Q_b = \langle \sum_i \left(\frac{n_i}{\sum_j n_j}\right)^b\rangle \approx \frac{\tau-T_c}{T_c(b-1)} $ -
All other merger rates are also consistent with the Bolthausen-Sznitman coalescent:
$\quad\lambda_{b,k} = \frac{(k-2)!(b-k)!}{T_c (b-1)!}$
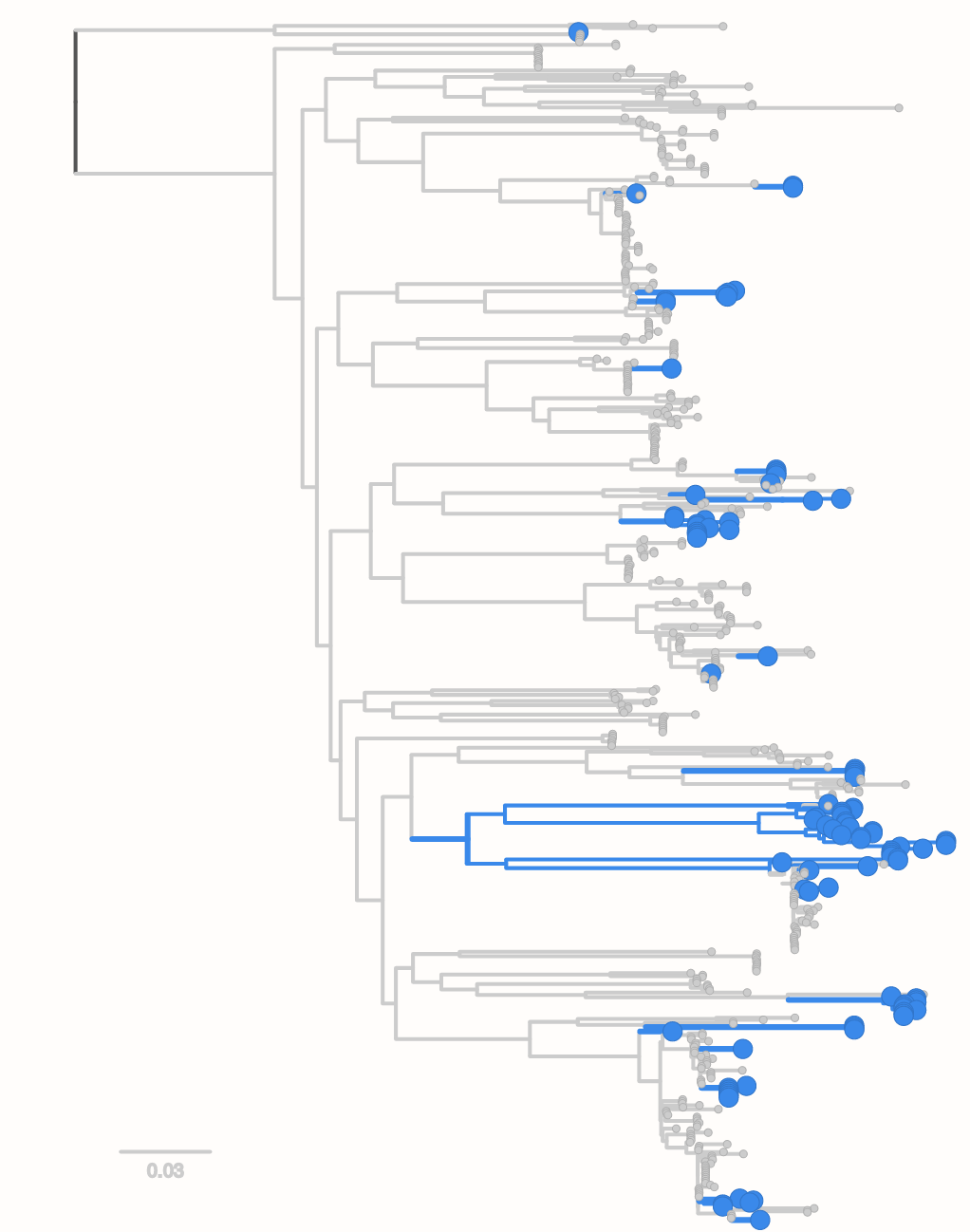
Bursts in a tree ↔ high fitness genotypes
Predicting evolution
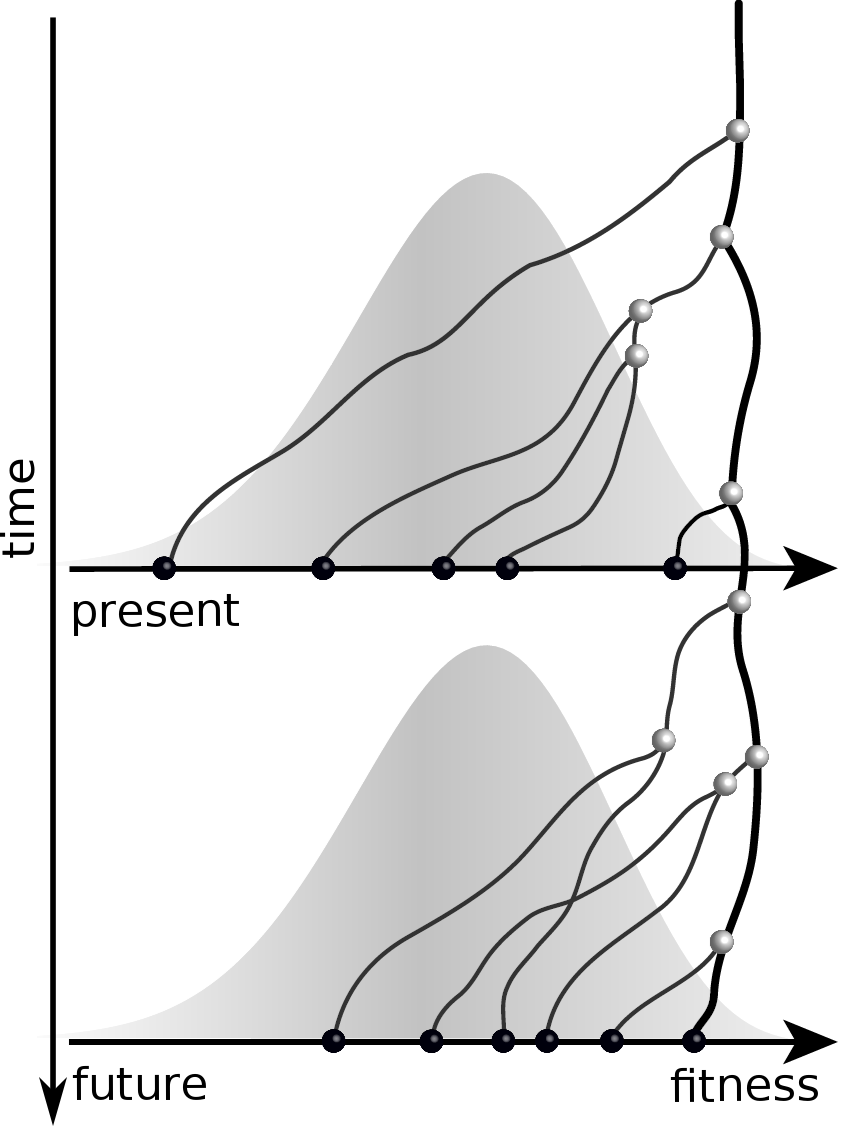
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
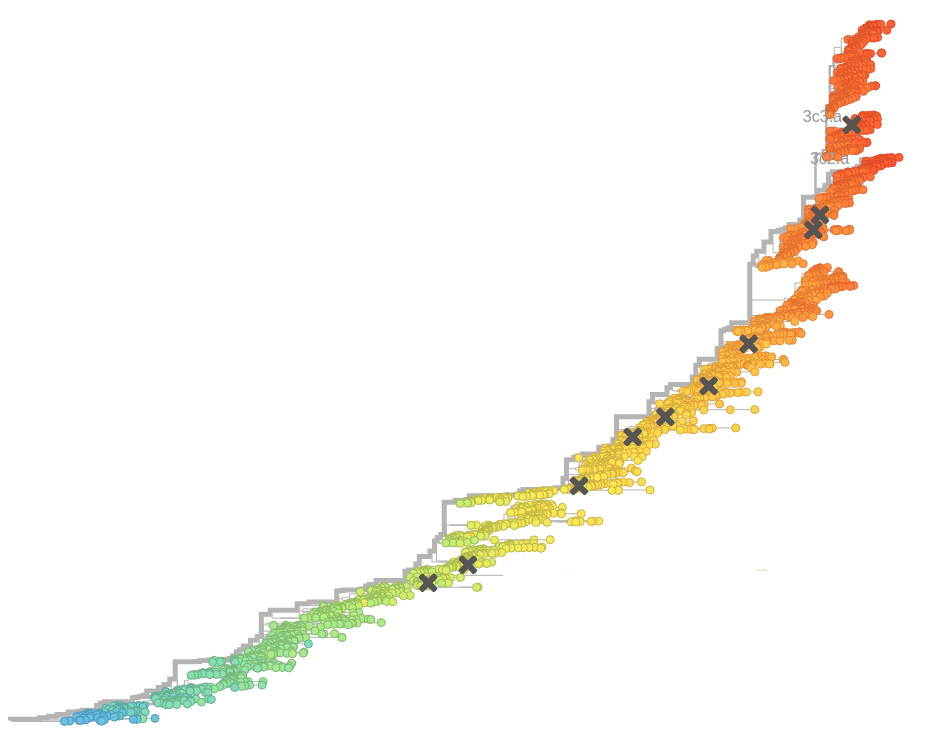
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
Prediction of the dominating H3N2 influenza strain
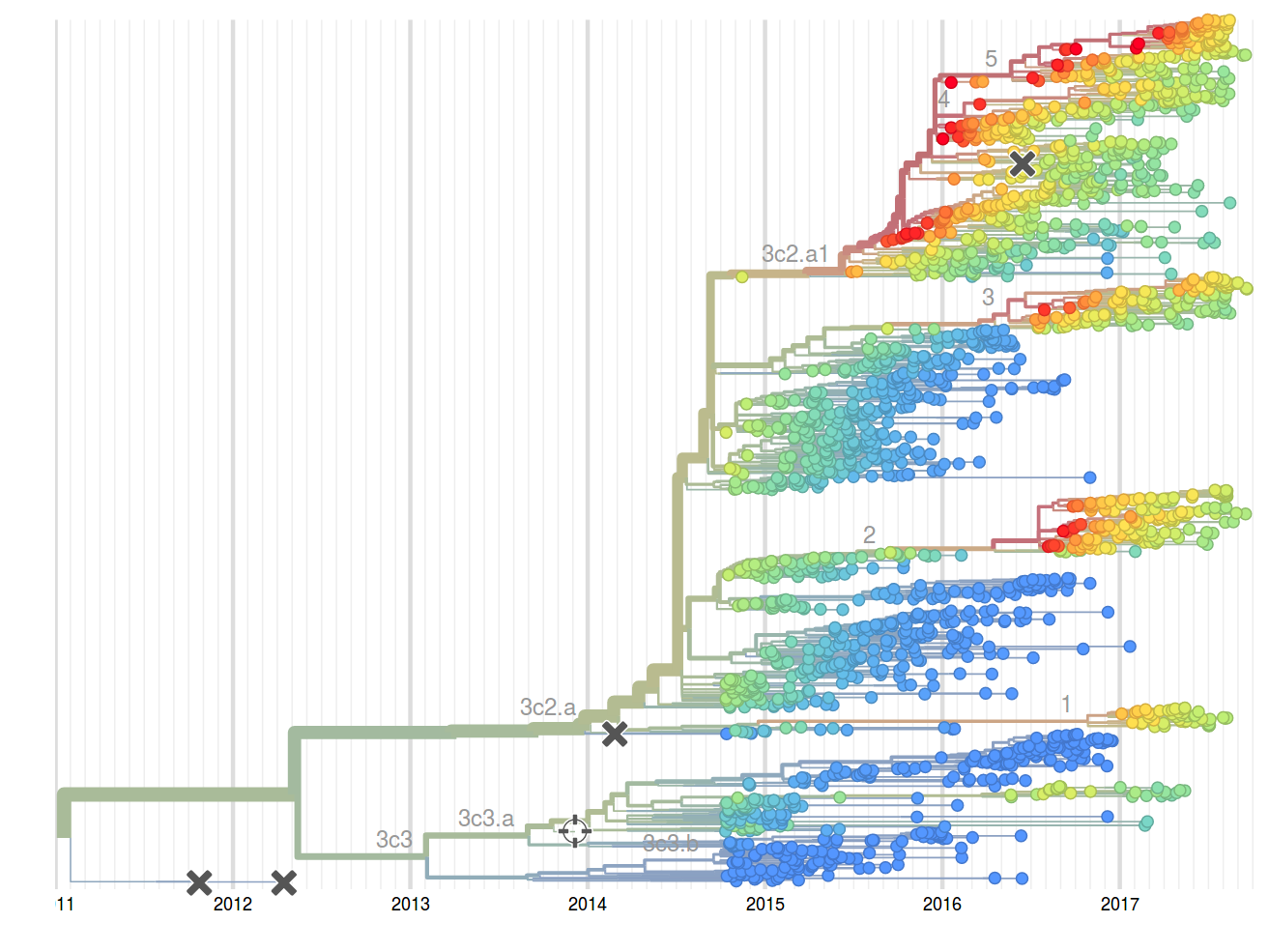
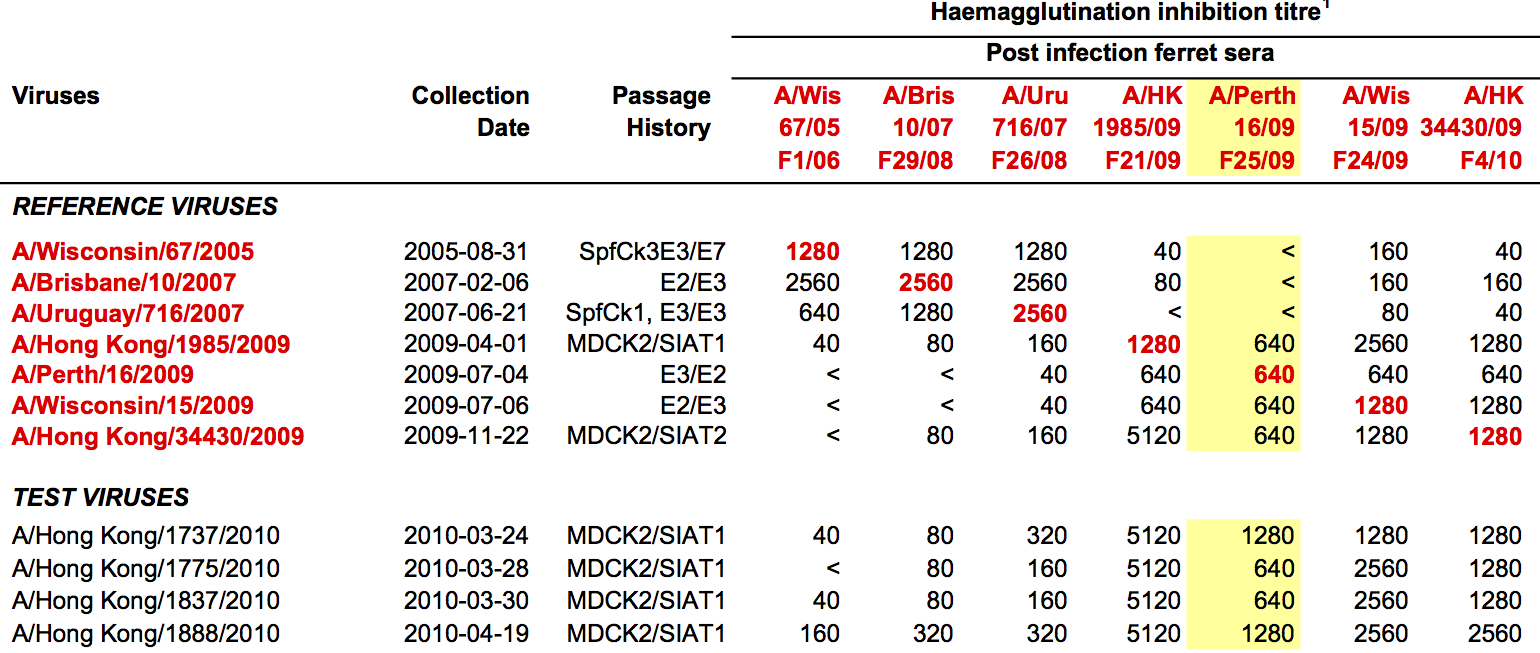
Hemagglutination Inhibition assays
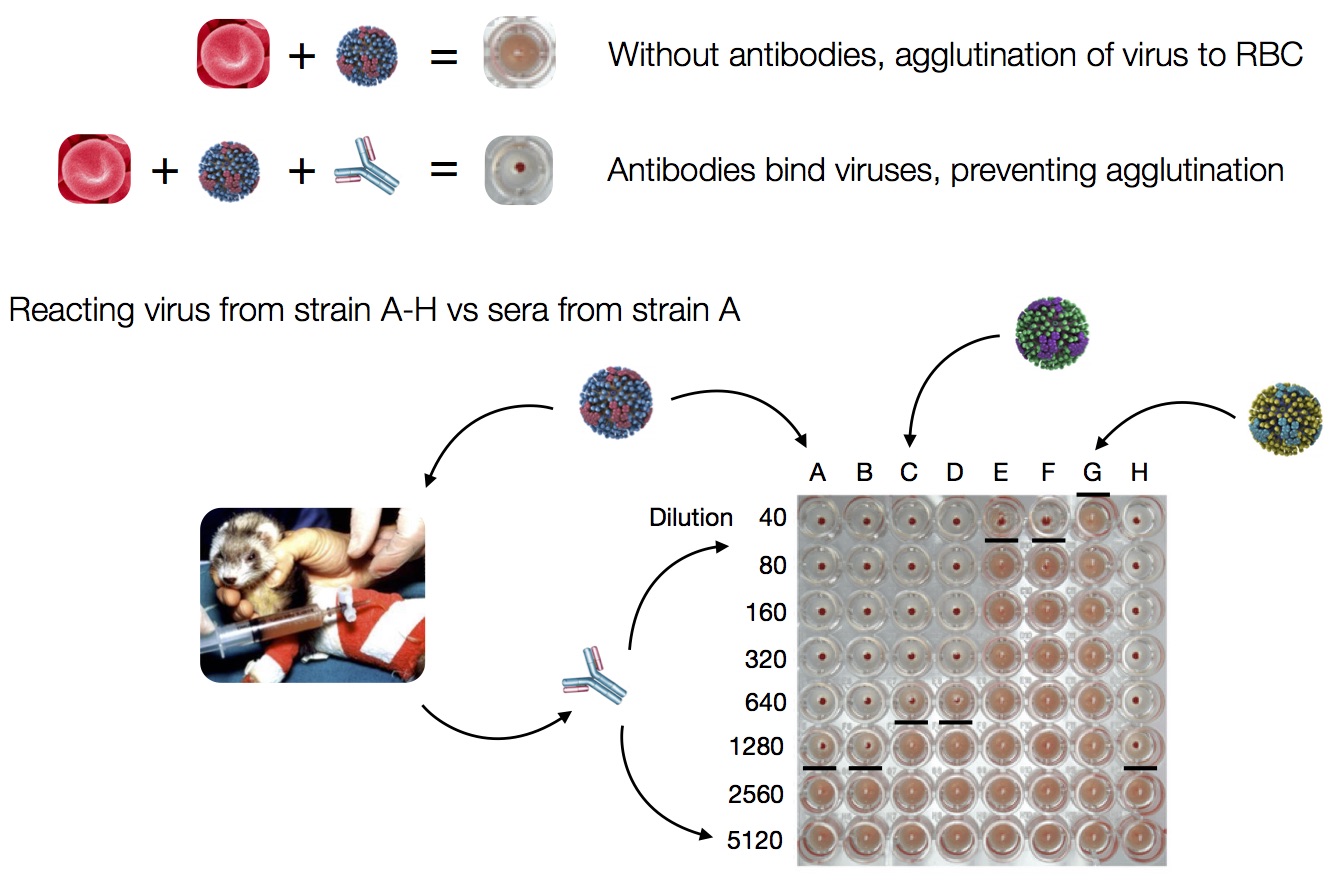
HI data sets
- Long list of distances between sera and viruses
- Tables are sparse, only close by pairs
- Structure of space is not immediately clear
- MDS in 2 or 3 dimensions
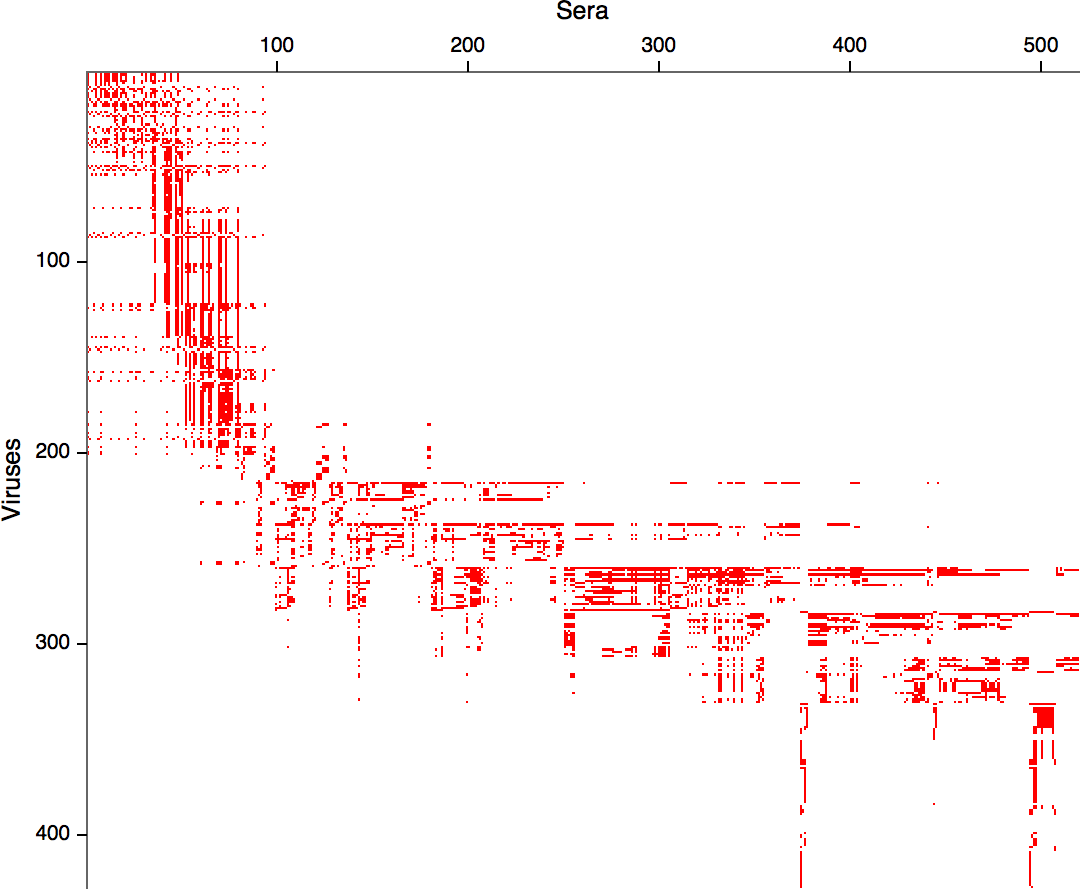
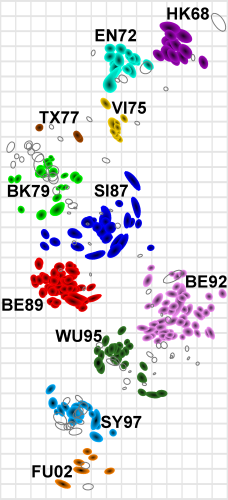 Smith et al, Science 2002
Smith et al, Science 2002
Integrating antigenic and molecular evolution

- $H_{a\beta} = v_a + p_\beta + \sum_{i\in (a,b)} d_i$
- each branch contributes $d_i$ to antigenic distance
- sparse solution for $d_i$ through $l_1$ regularization
Integrating antigenic and molecular evolution

- MDS: $(d+1)$ parameters per virus
- Tree model: $2$ parameters per virus
- Sparse solution
→ identify branches or substitutions that cause titer drop
but...
- HI titers are a crude proxy for selection in humans
- need human serology + infection history
- better coverage where most of influenza infections happen
Lots of reassortement
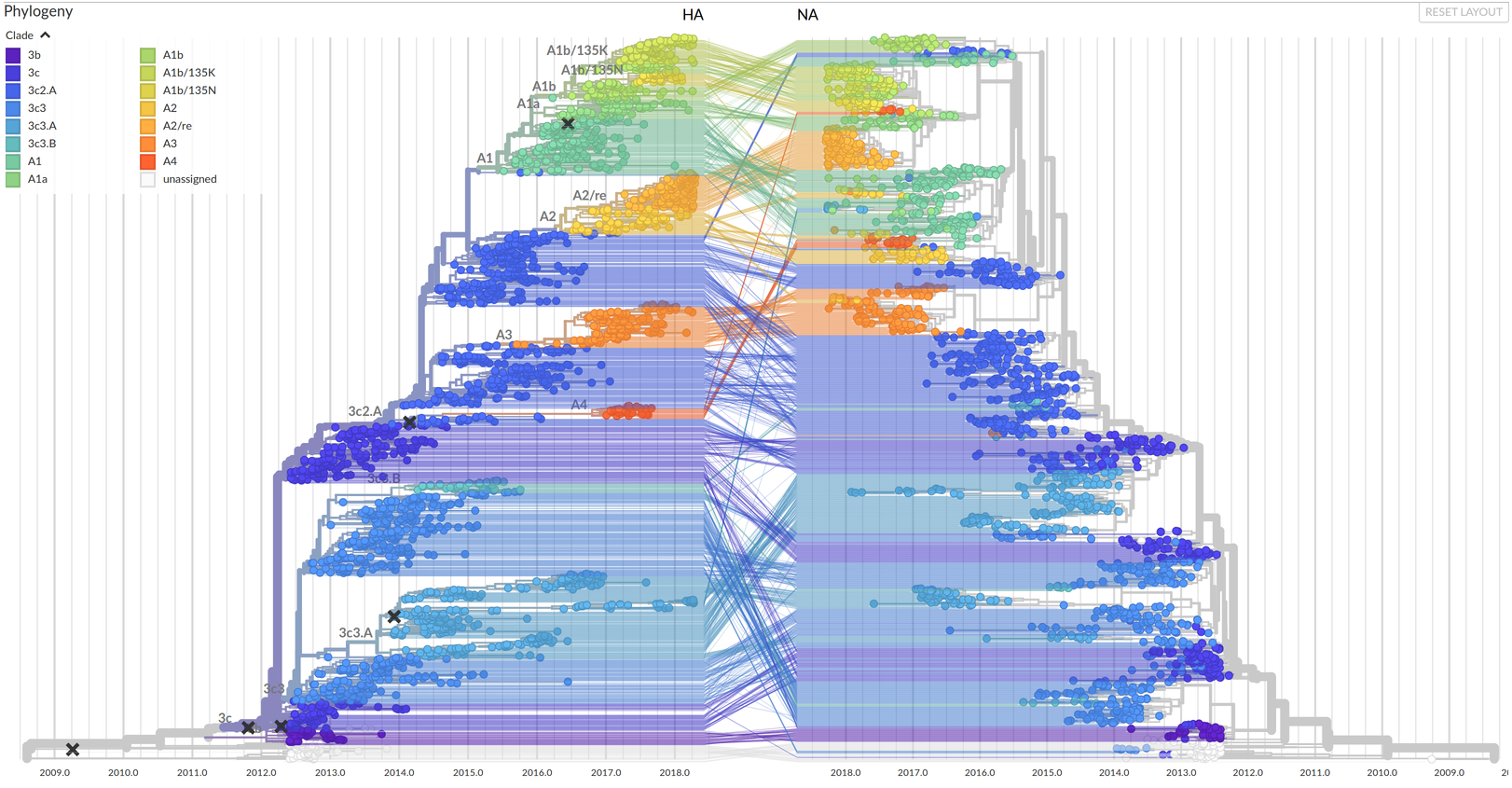
next to no idea what mutations in segments other than HA do...
What about bacteria?
- vertical and horizontal transmission
- genome rearrangements
- much larger genomes
- NGS genomes tend to be fragmented
- very little understanding of relevant phenotypes
- what are the relevant spatial and temporal scales?

panX by Wei Ding
- pan-genome identification pipeline
- phylogenetic analysis of each orthologous cluster
- detect associations with phenotypes
- fast: analyze hundreds of genomes in a few hours
- github.com/neherlab/pan-genome-analysis
Bacteria: Species trees and gene trees
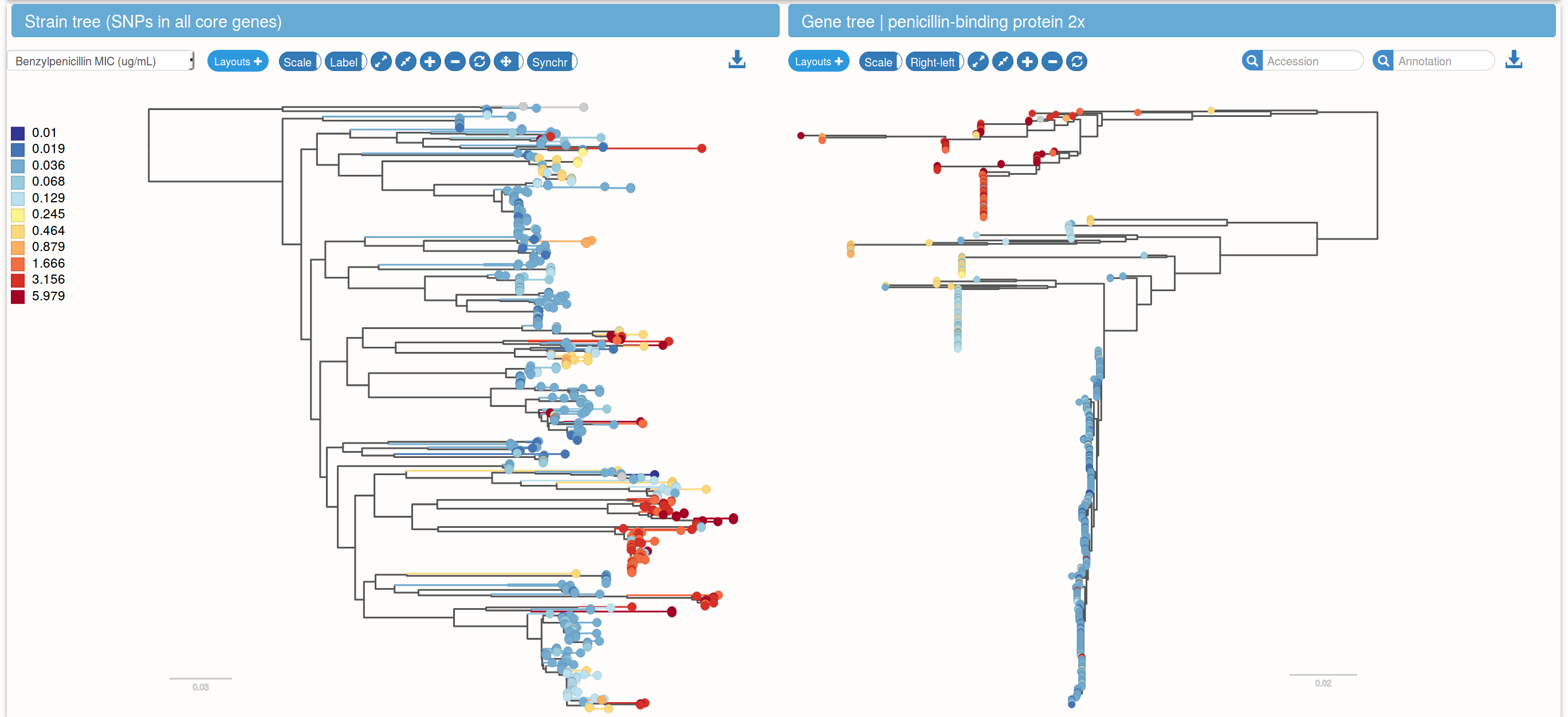
Structural variation
- closely related strains differ in dozens to hundreds of genes
- frequent rearrangements/loss of synteny
- often multiple plasmids from a few to several 100 thousand bases
- recombination necessary for genome maintainance and segregation
- most evolutionary analyses look for SNPs in a "core genome"
- but the action is somewhere else...
Genome assembly with short reads

- 10s of millions of short reads (<500bp)
- Too short to bridge repetitive elements
- → assemblies are fragmented into 100s of "contigs"
(really terrible example)
Long-read sequencing



Ancestral recombination graphs
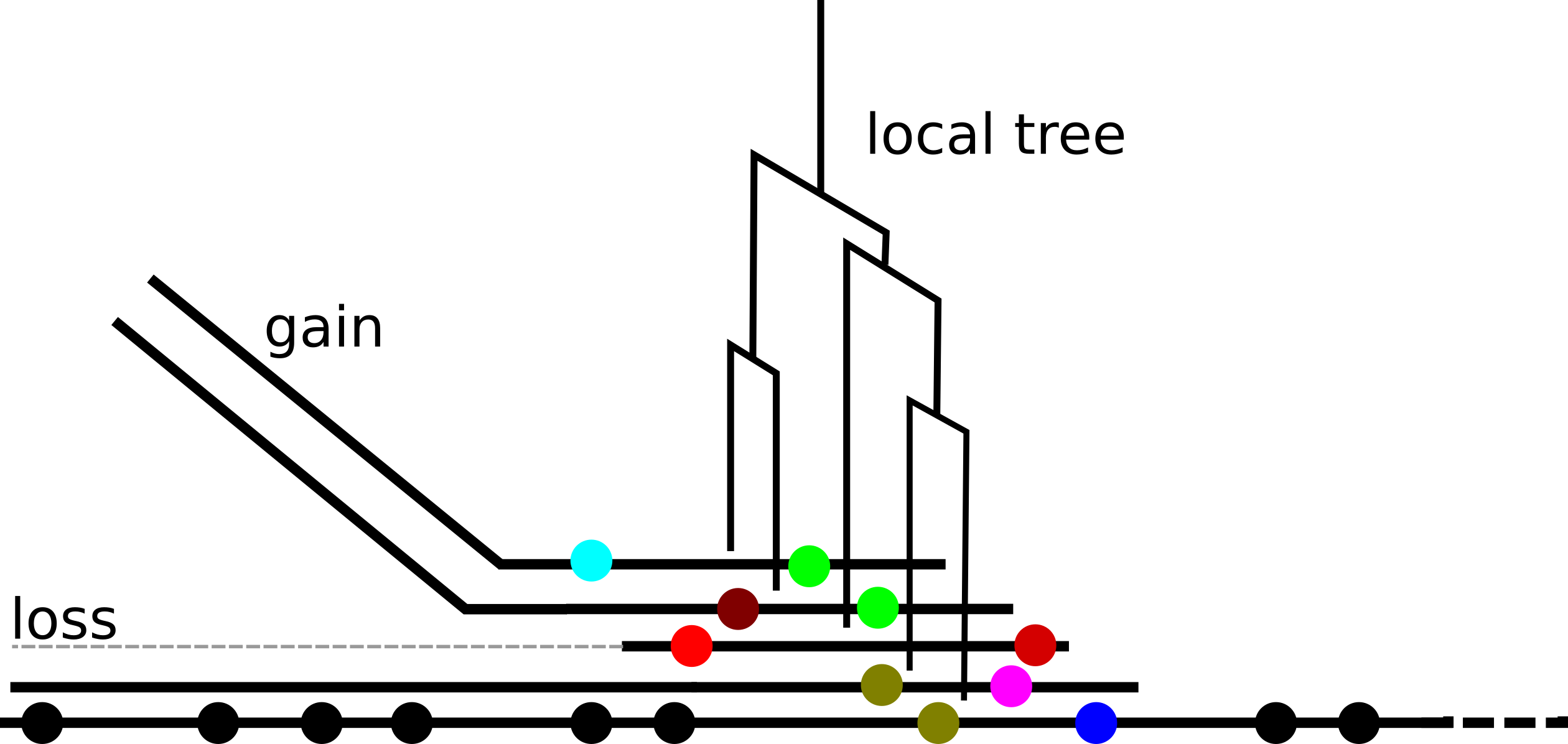
- find orthologous clusters → use as seeds for local alignments
- stitch together into a larger graph
- identify synteny blocks, recombination hotspots, across species transfers...
Another global experiment...