Tracking pathogenic bacteria and AMR at the plasmid, genome, and pan-genome level
Nicholas Noll & Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/201811_kyoto.html
Tracking pathogens is straightforward... if they are RNA viruses
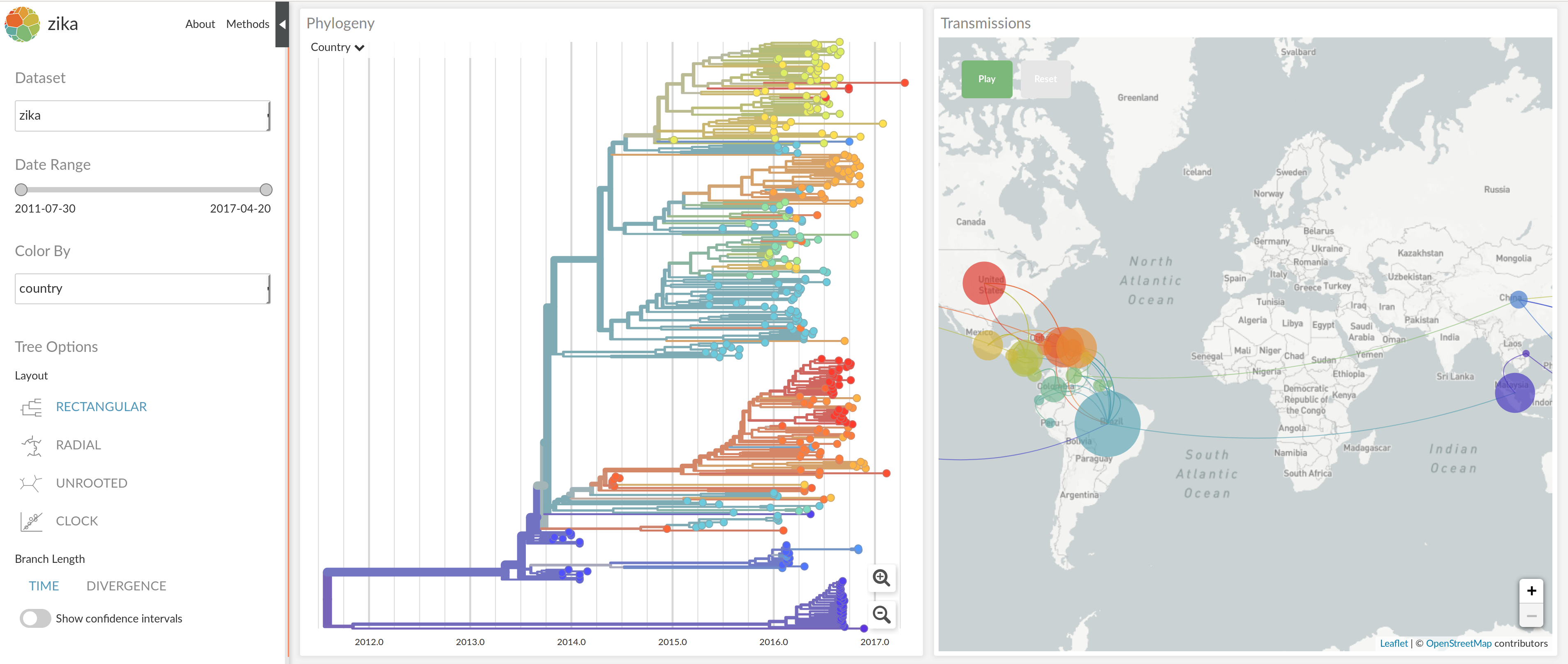
What genetic unit do we track?
What are good ways of visualizing bacterial diversity?
What are meaningful distance measures?
Why is this hard?
- every genome contains a different set of genes
- horizontal transfer
- genome rearrangements
- variable divergence along the genome
- important parts sit on plasmids,
flanked by repetitive sequence - short read assemblies tend to be fragmented
- annotations of variable quality

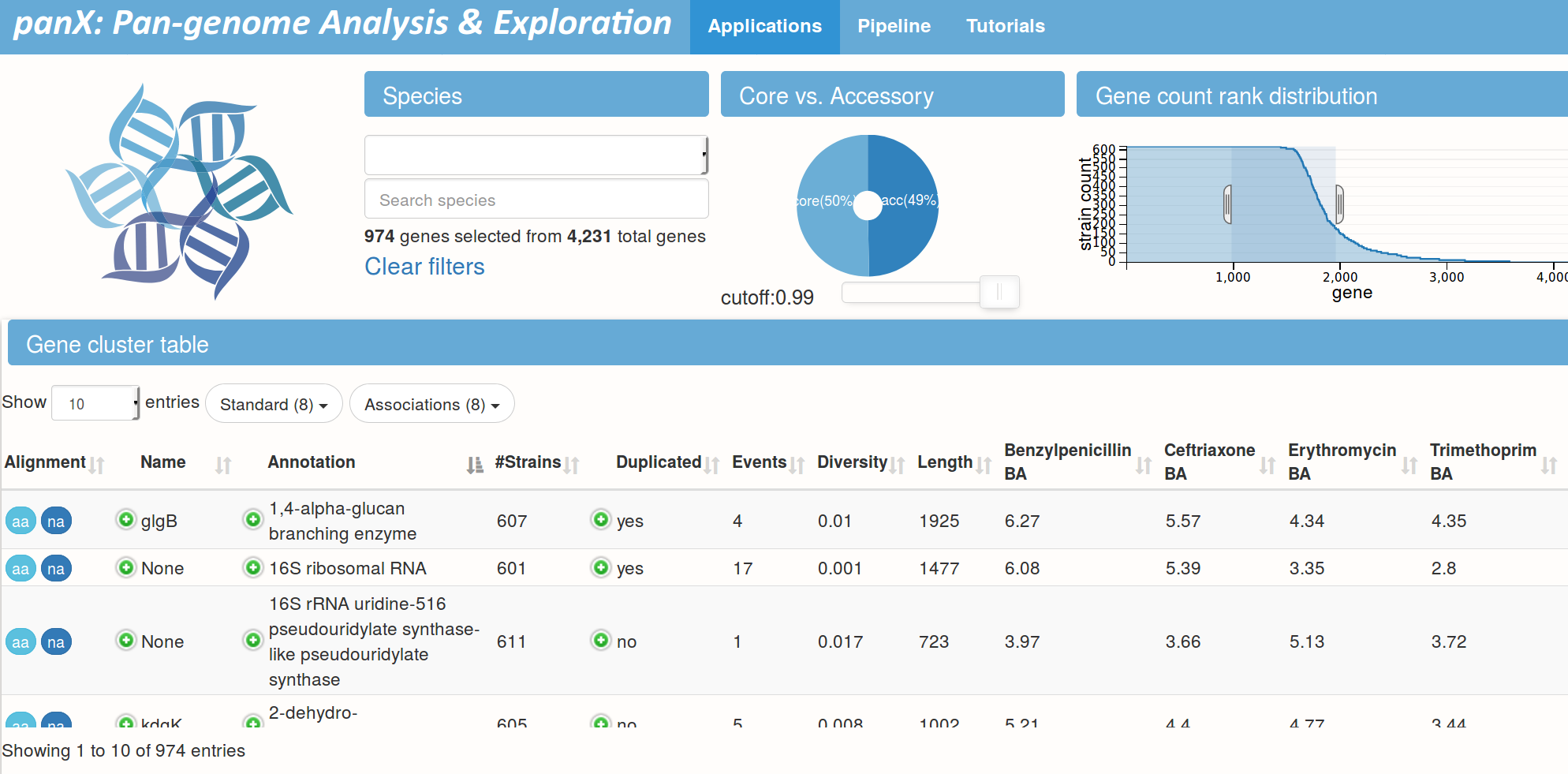
panX by Wei Ding
- pan-genome identification pipeline
- phylogenetic analysis of each orthologous cluster
- detect associations with phenotypes
- fast: analyze hundreds of genomes in a few hours
- github.com/neherlab/pan-genome-analysis
panX @ pangenome.de
S. pneumoniae data set by Croucher et al.
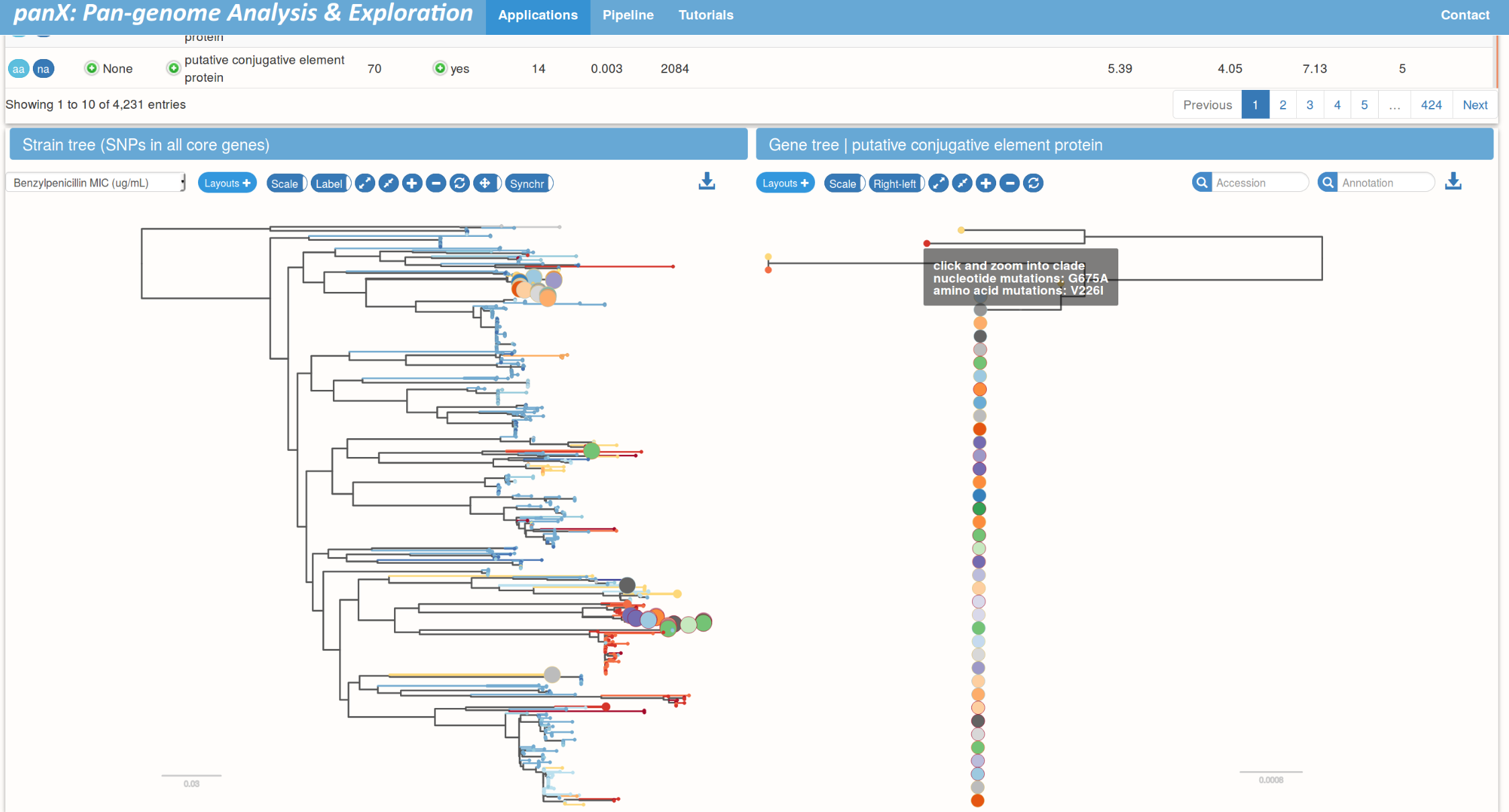
Pan-genome statistics and filters
Species trees and gene trees
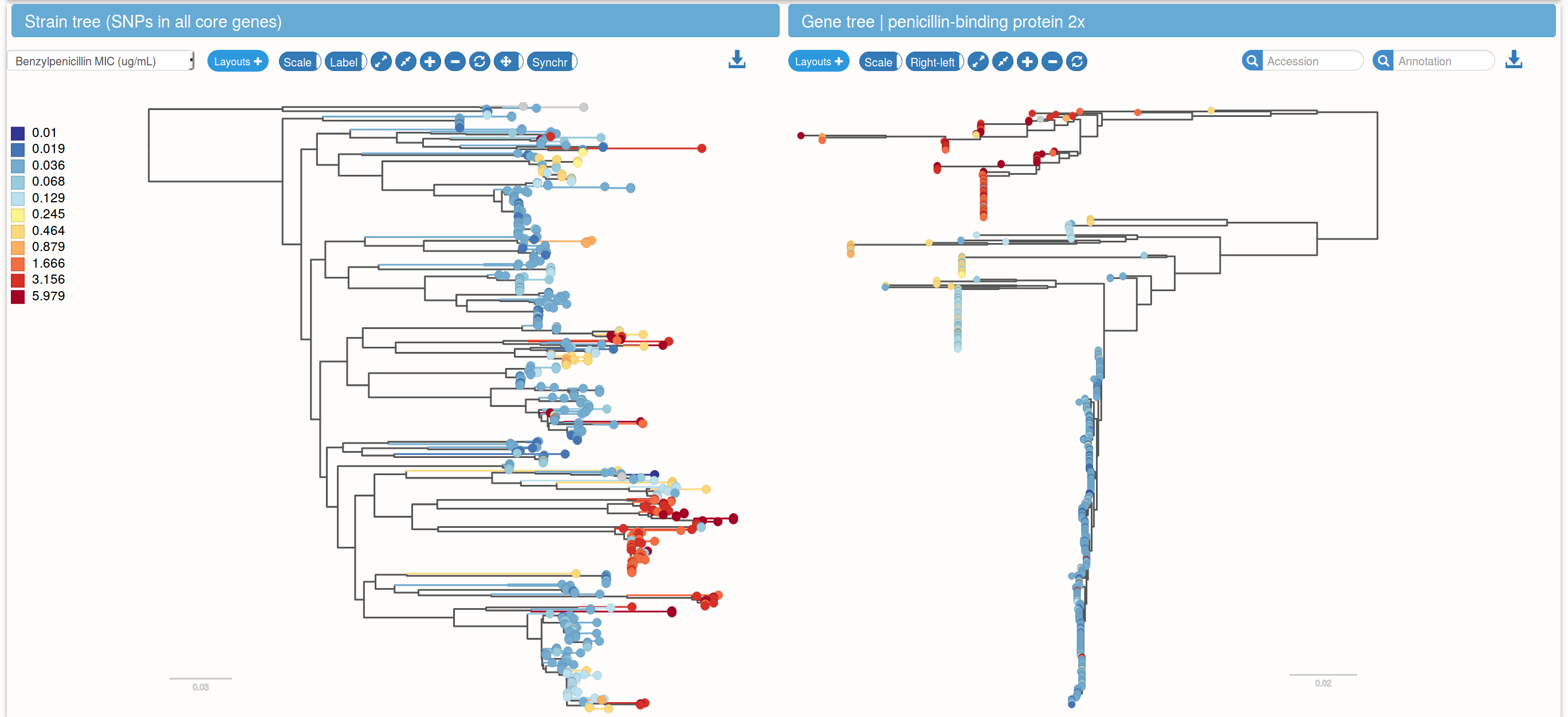
Links between species trees and gene trees
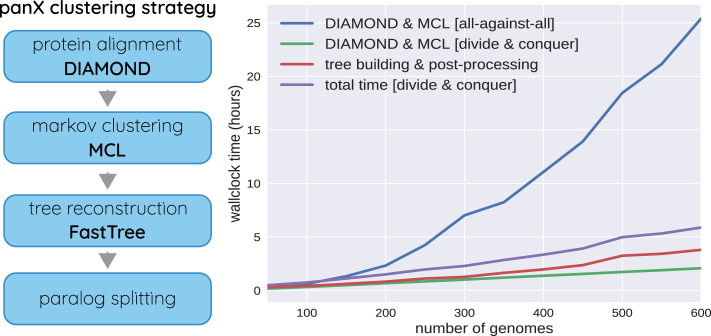
- make pan-genomes in batches of 50
- construct pseudo-consensus genomes for each batch
- cluster the batch consensi
- expand clusters
Cluster aggressively, split paralogs later
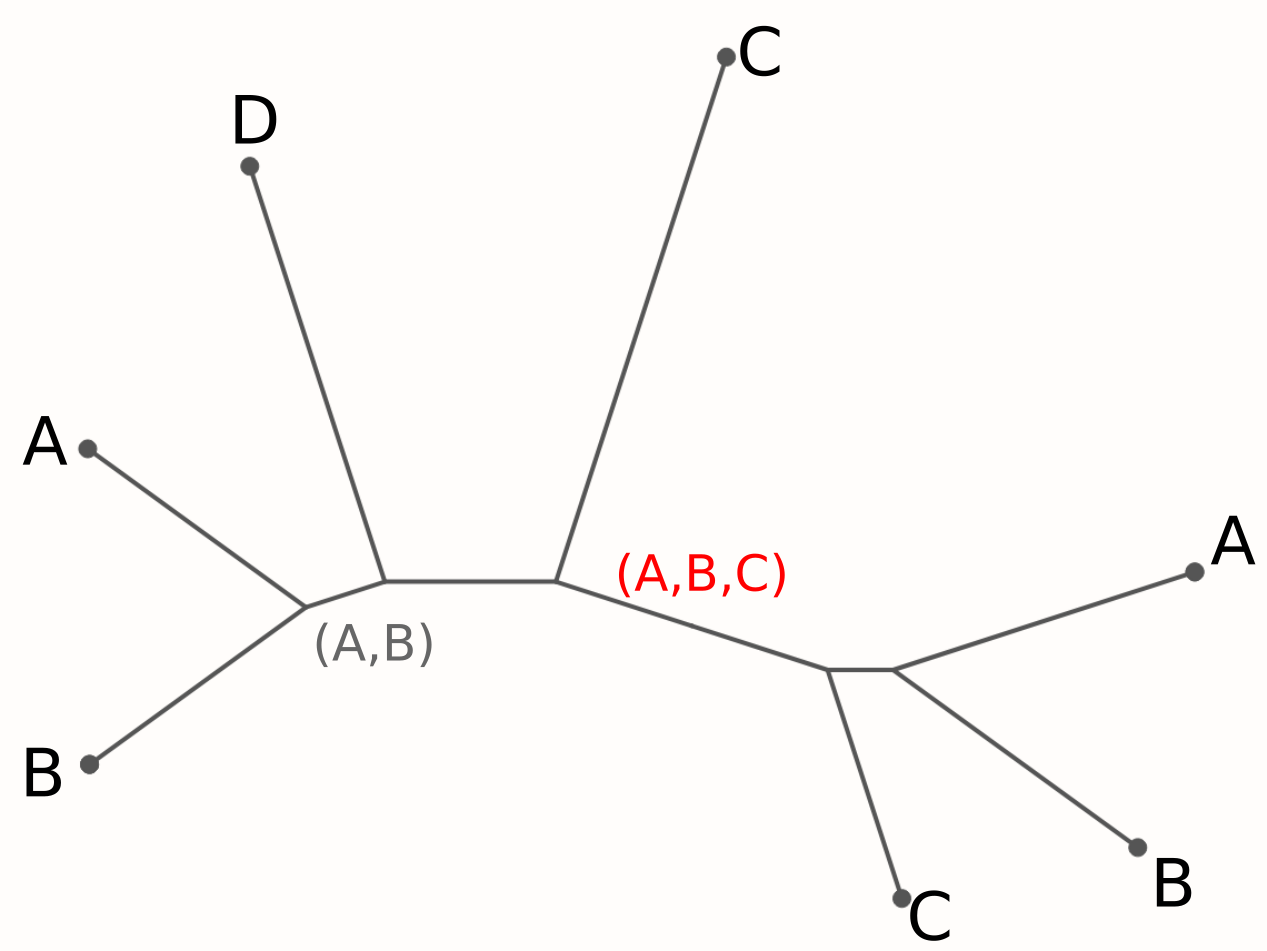
Find biggest intersection of left/right leaves.
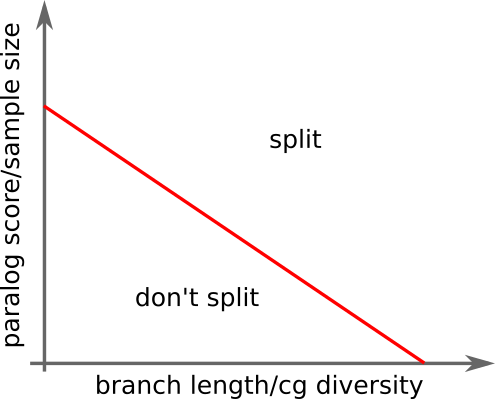
cut branch depending on paralog score.
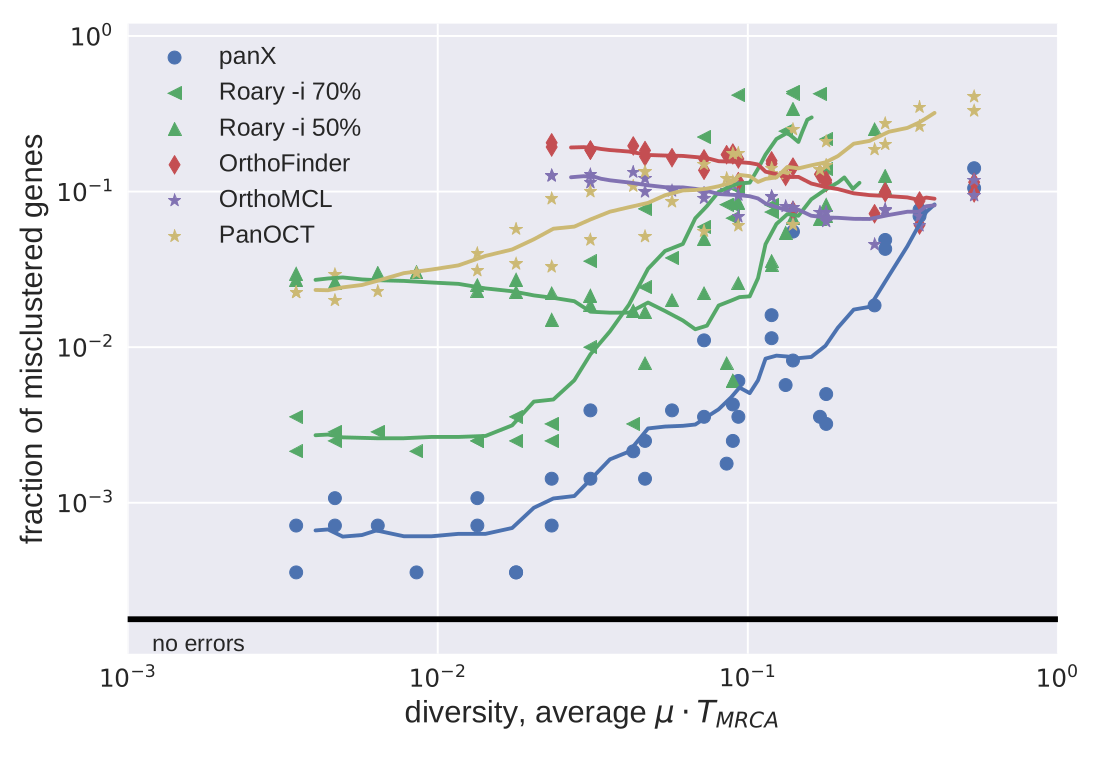
Ortholog cluster accuracy
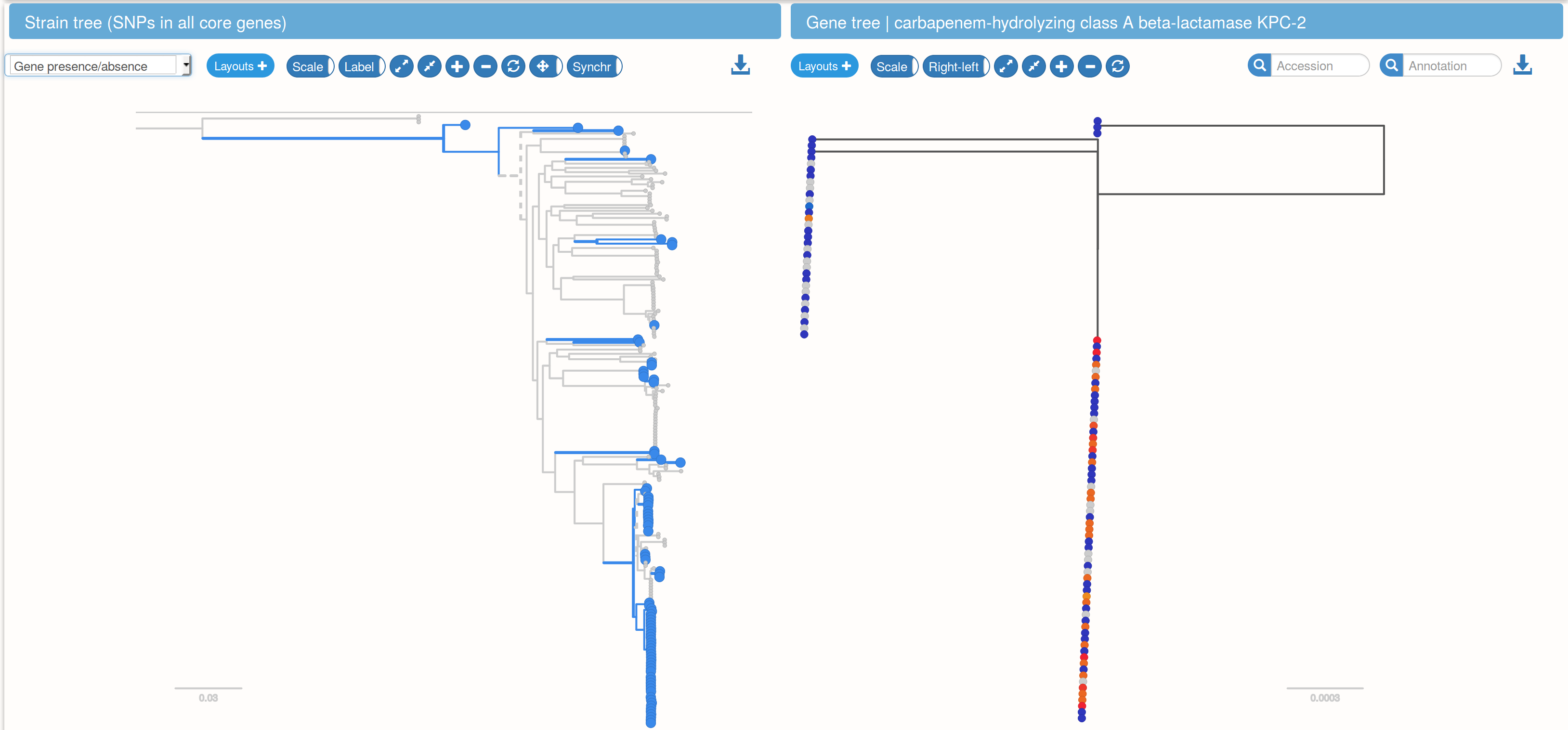
Mobile AMR genes have provide little phylogenetic resolution (example KPC)