Using open data to track and predict infectious disease
Richard Neher
Biozentrum & SIB, University of Basel
slides at neherlab.org/201910_RIVM.html
Sequences record the spread of pathogens
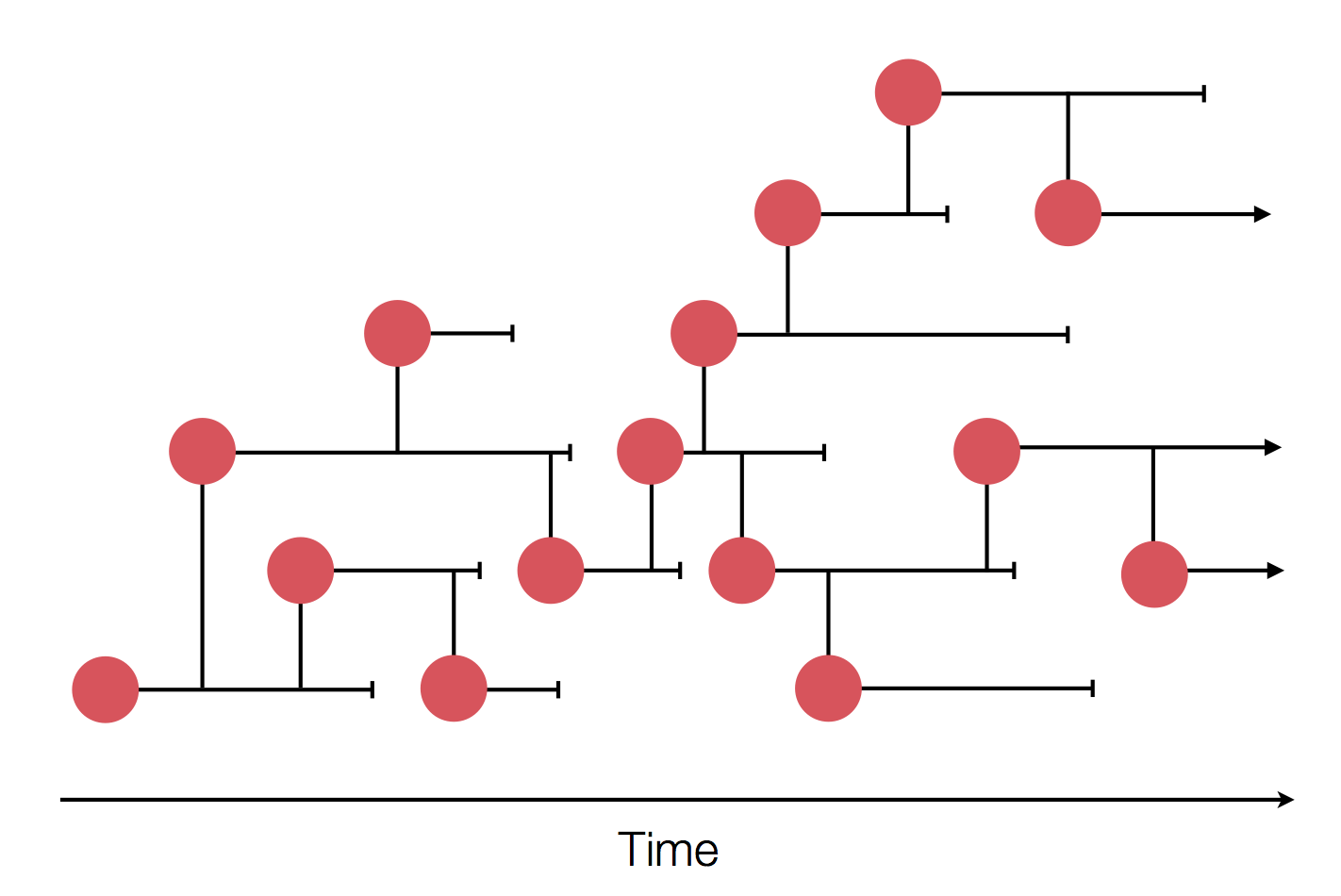
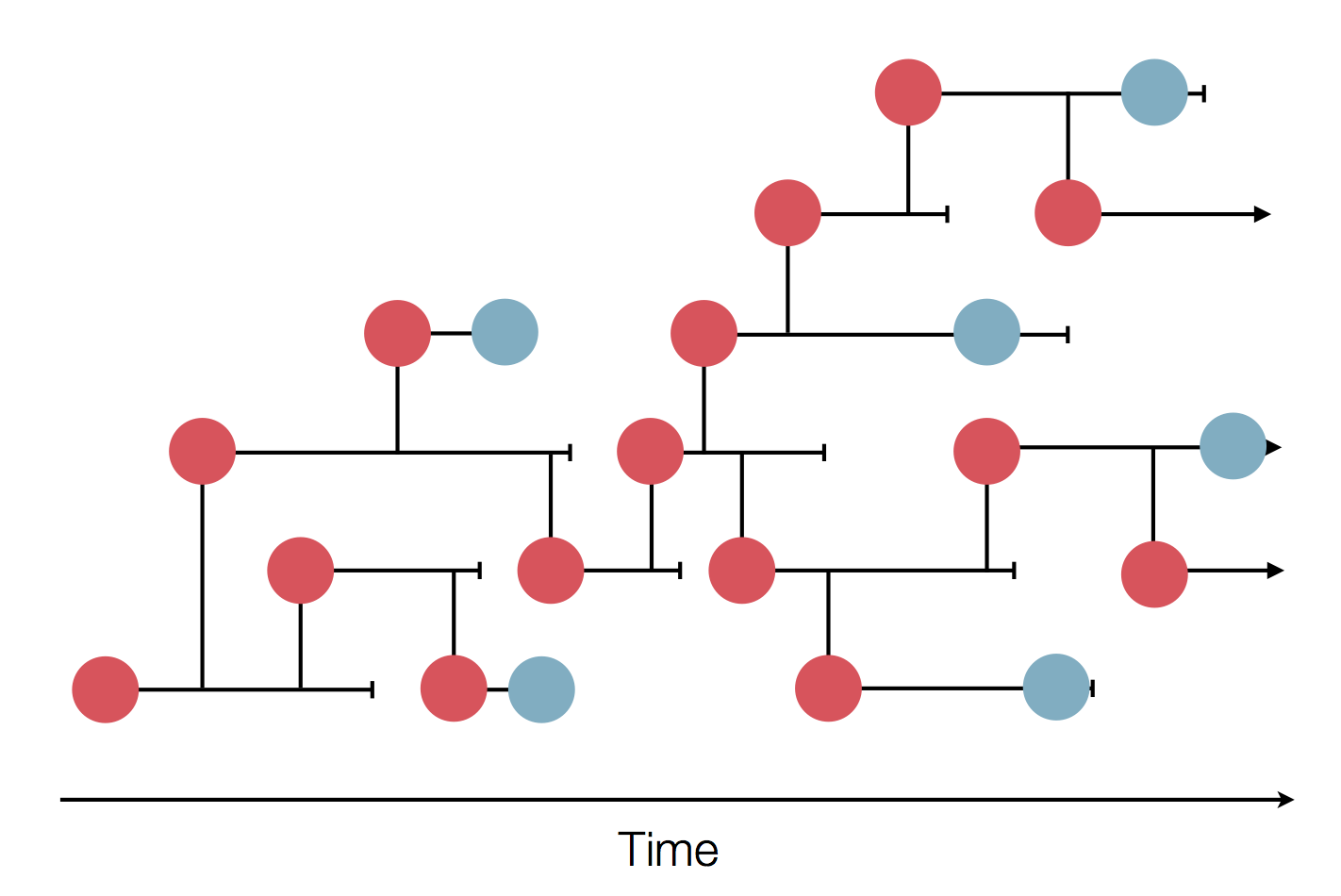
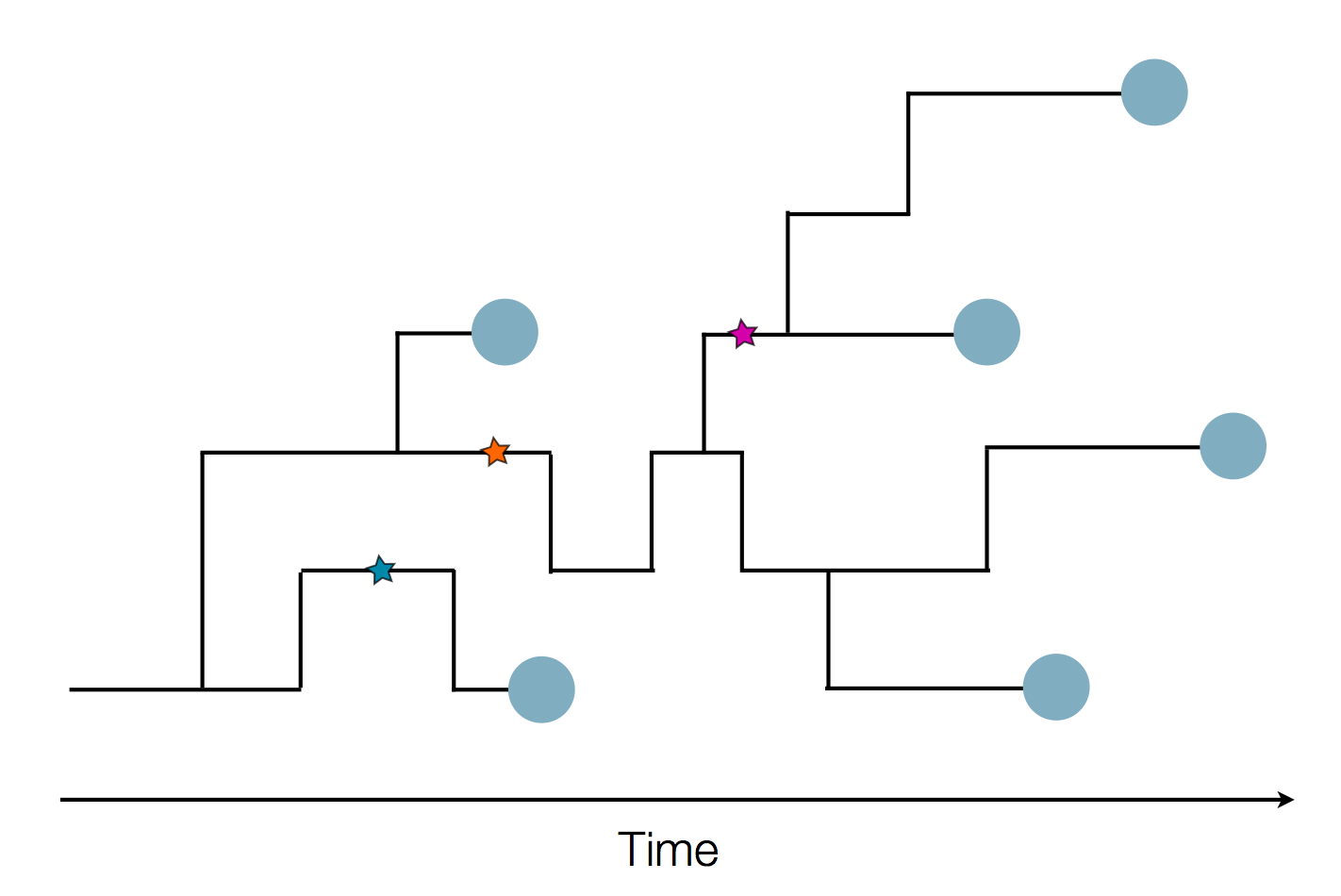
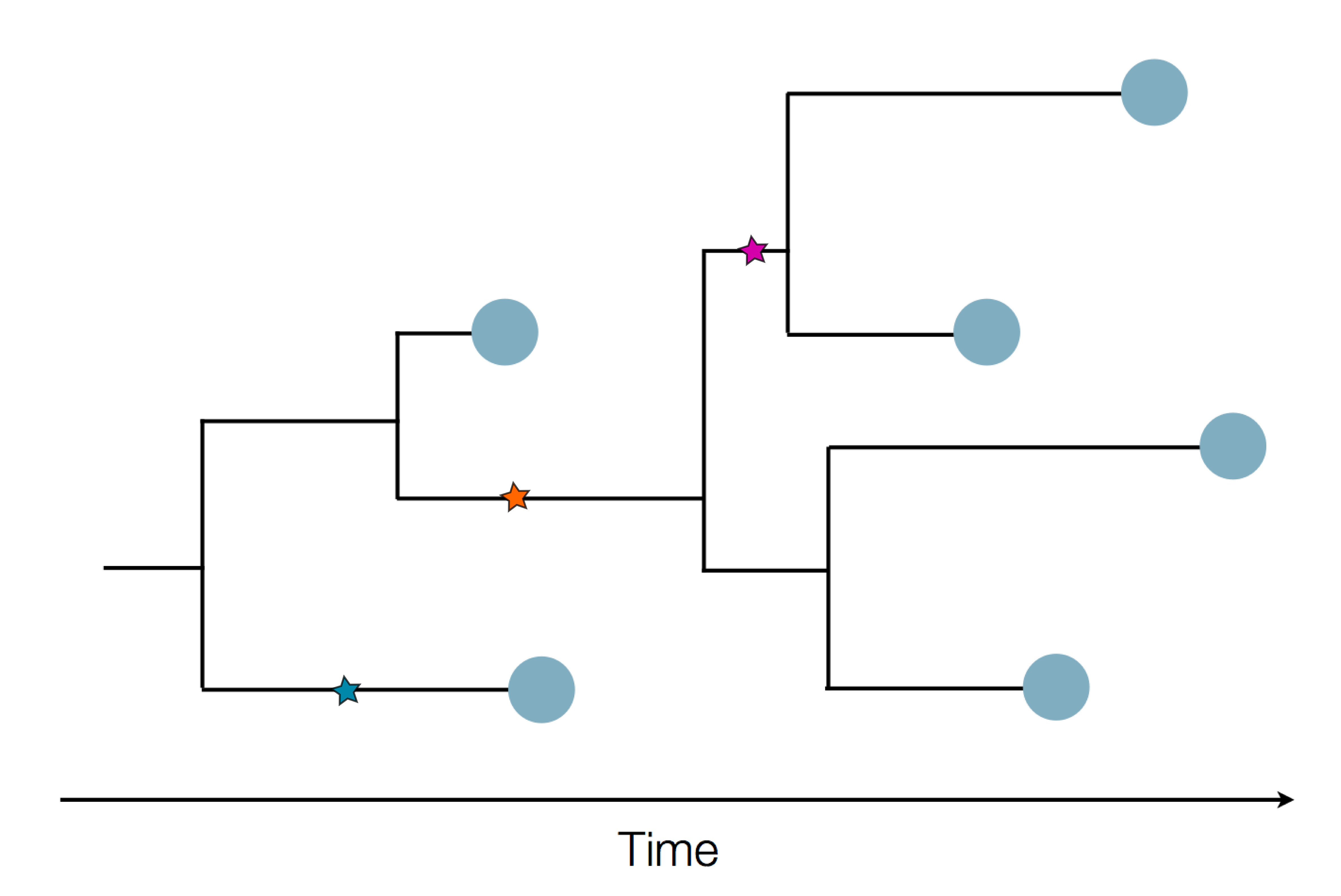
Mutations accumulate at a rate of $10^{-5}$ per site and day!
Influenza virus genome - 8 segments
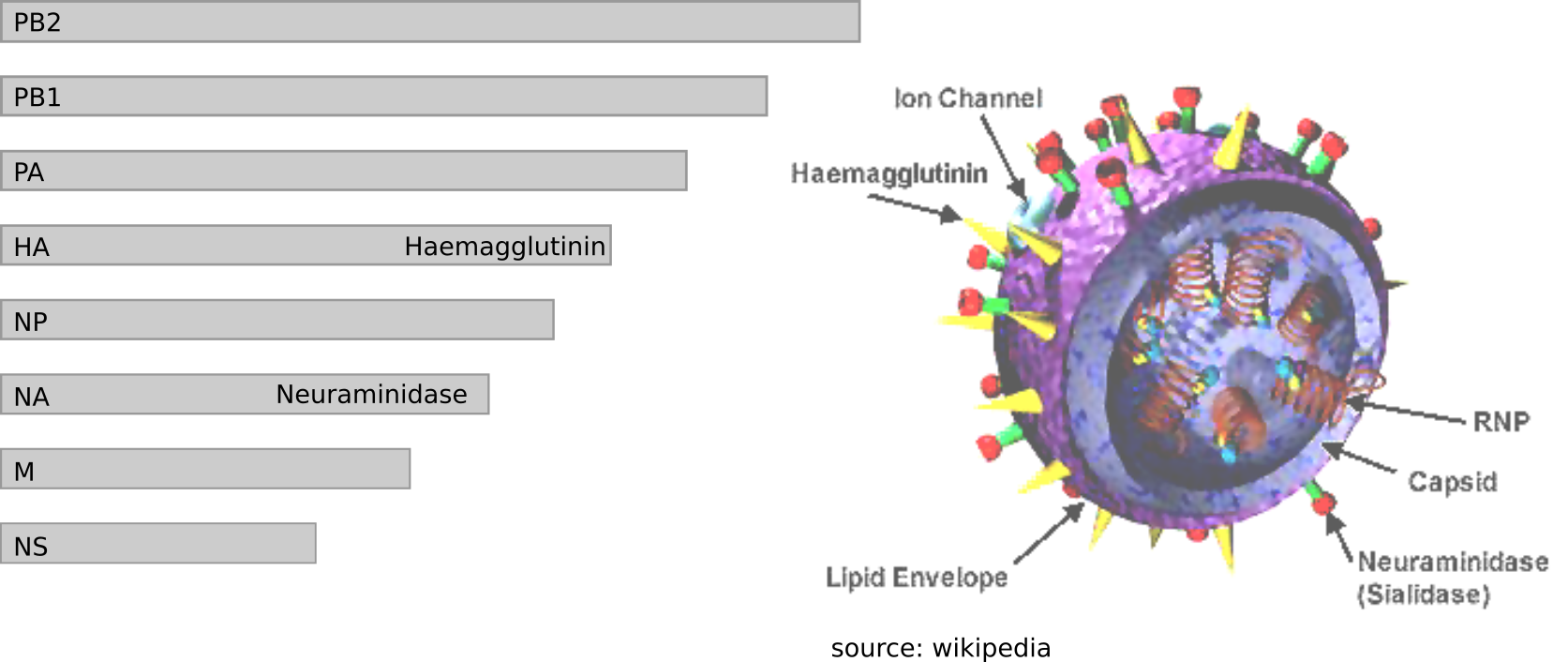
Zika virus genome $\sim 10000$ bases

Ebola virus genome $\sim 20000$ bases

Many RNA viruses pick up one mutation every 2-4 weeks!
Frequent mutations imply...
- most viruses in an outbreak/season differ from each other
- transmission chains are can be inferred
- transmission can be ruled out!
- geographic spread can be reconstructed
- drug resistance surveillance
- specific mutations might mediate antigenic mismatch
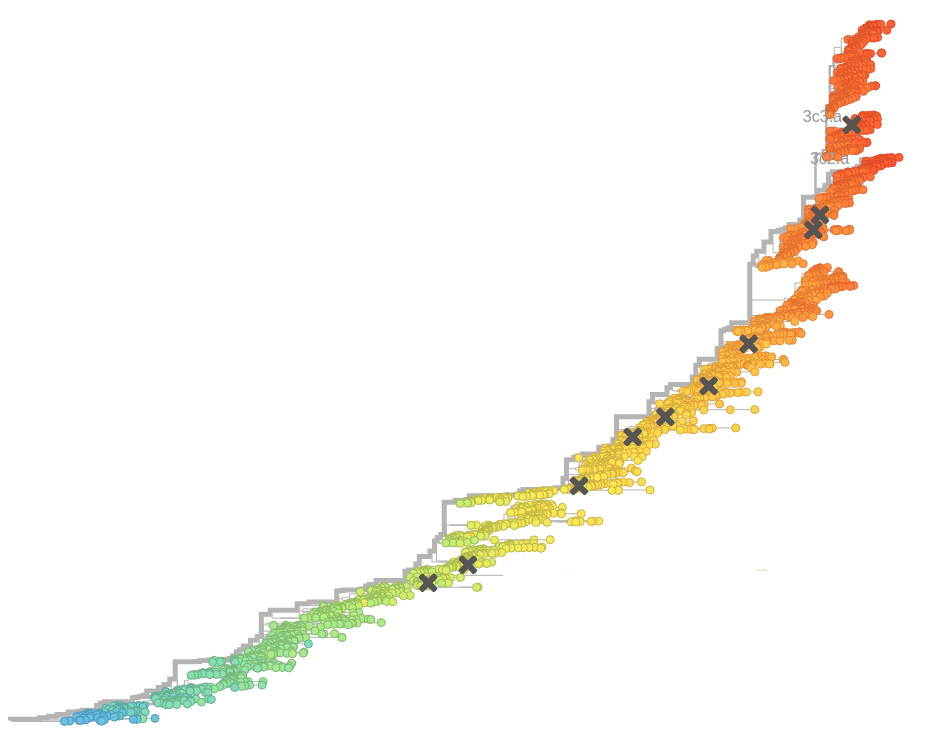
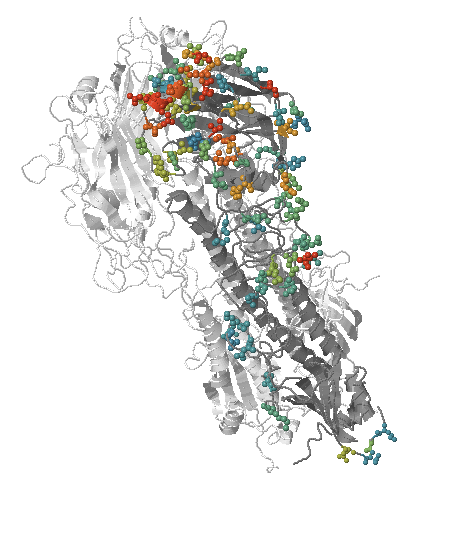
- Influenza viruses evolve to avoid human immunity
- Vaccines need frequent updates
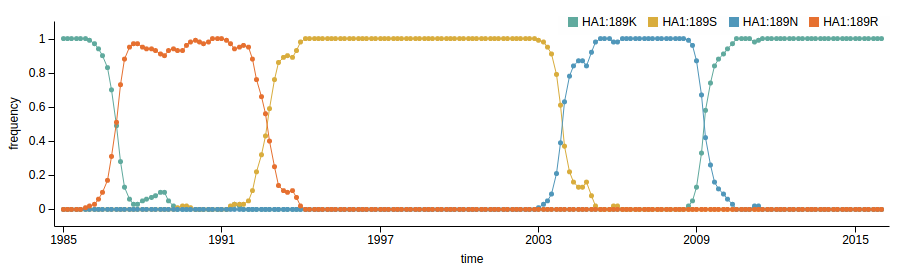
Vaccine strain selection schedule
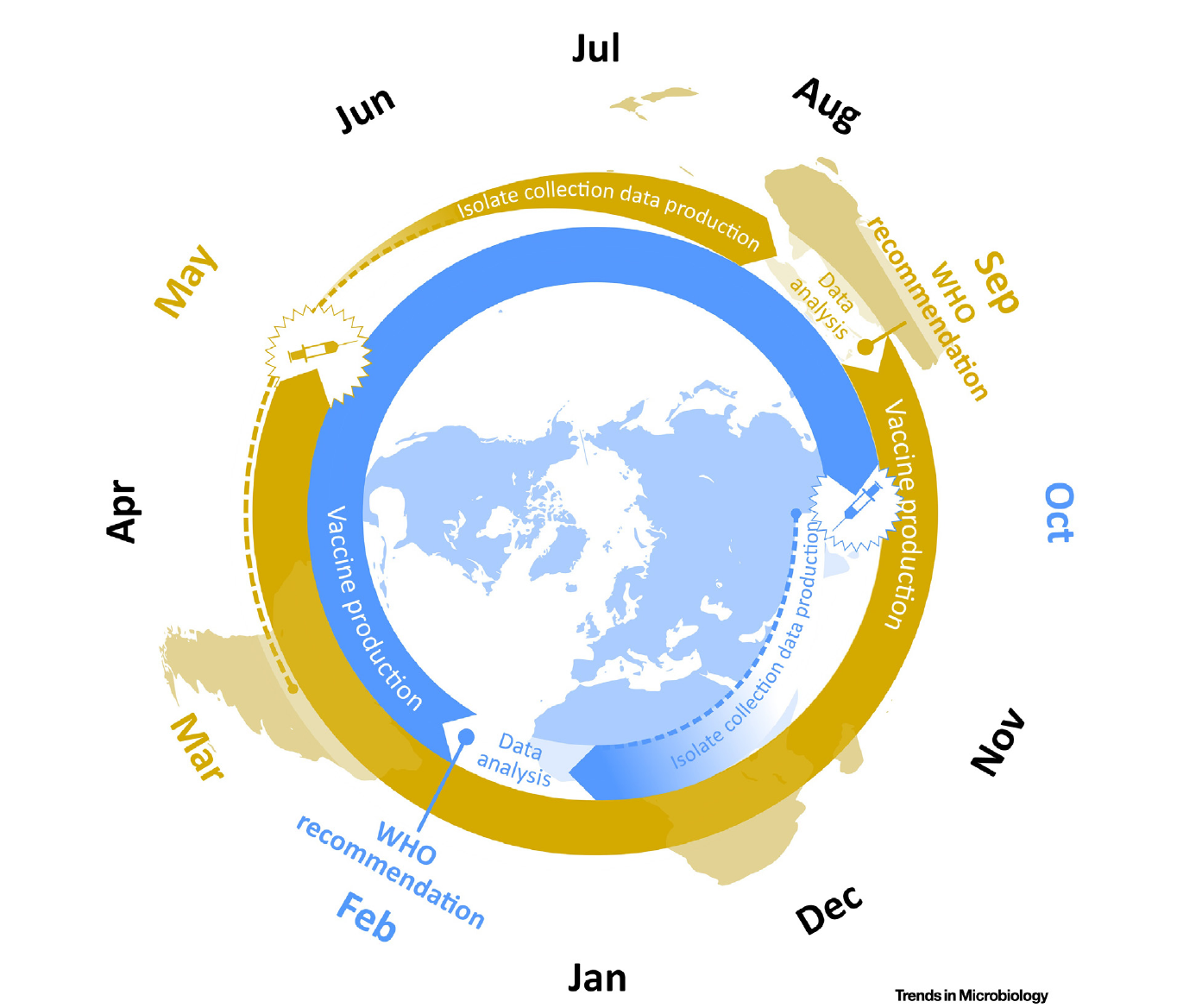 Klingen and McHardy, Trends in Microbiology
Klingen and McHardy, Trends in Microbiology
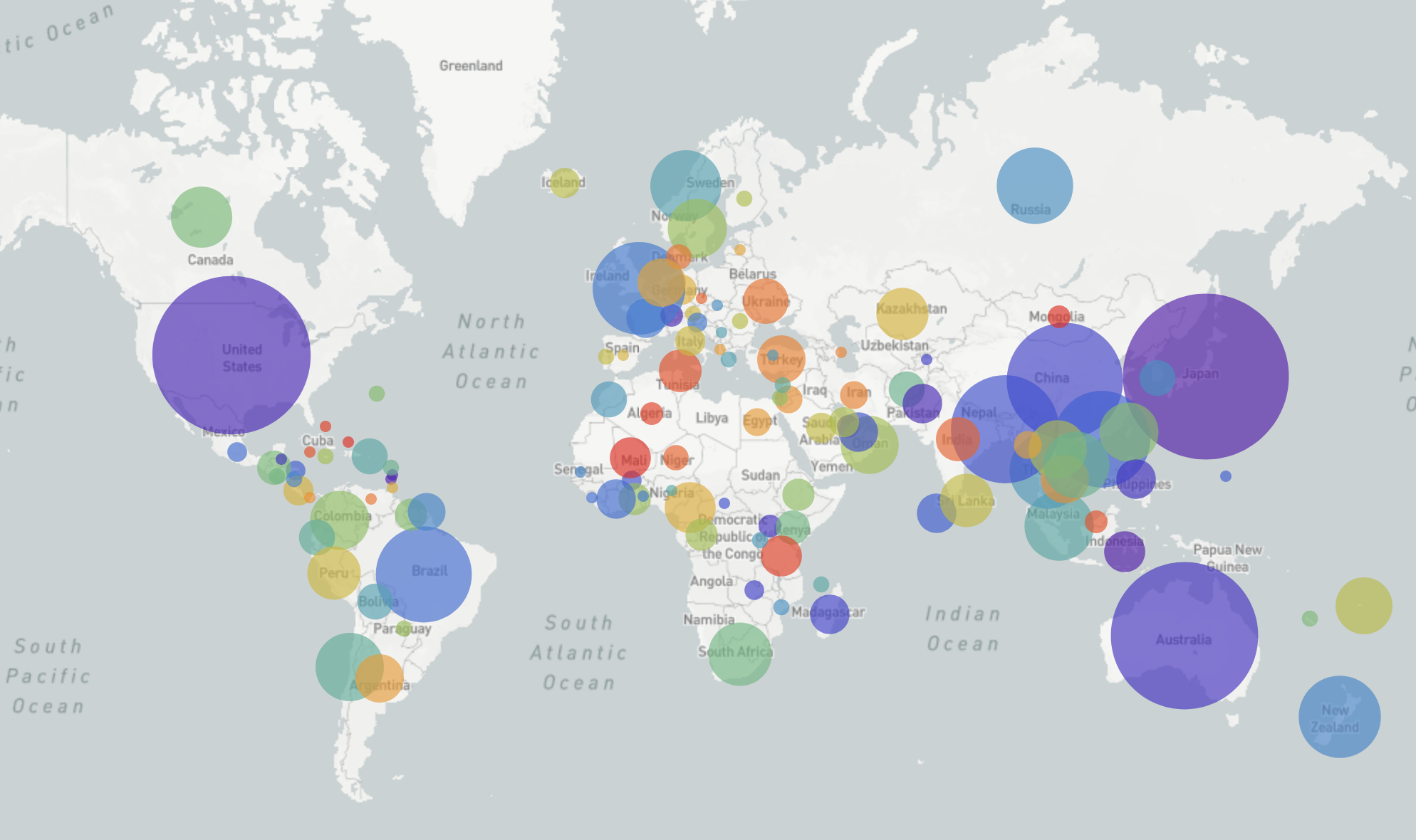
GISRS and GISAID -- Influenza virus surveillance
- comprehensive coverage of the world
- timely sharing of data through GISAID -- often within 2-3 weeks of sampling
- hundreds of sequences per week (in peak months)
→ requires continuous analysis and easy dissemination
→ interpretable and intuitive visualization
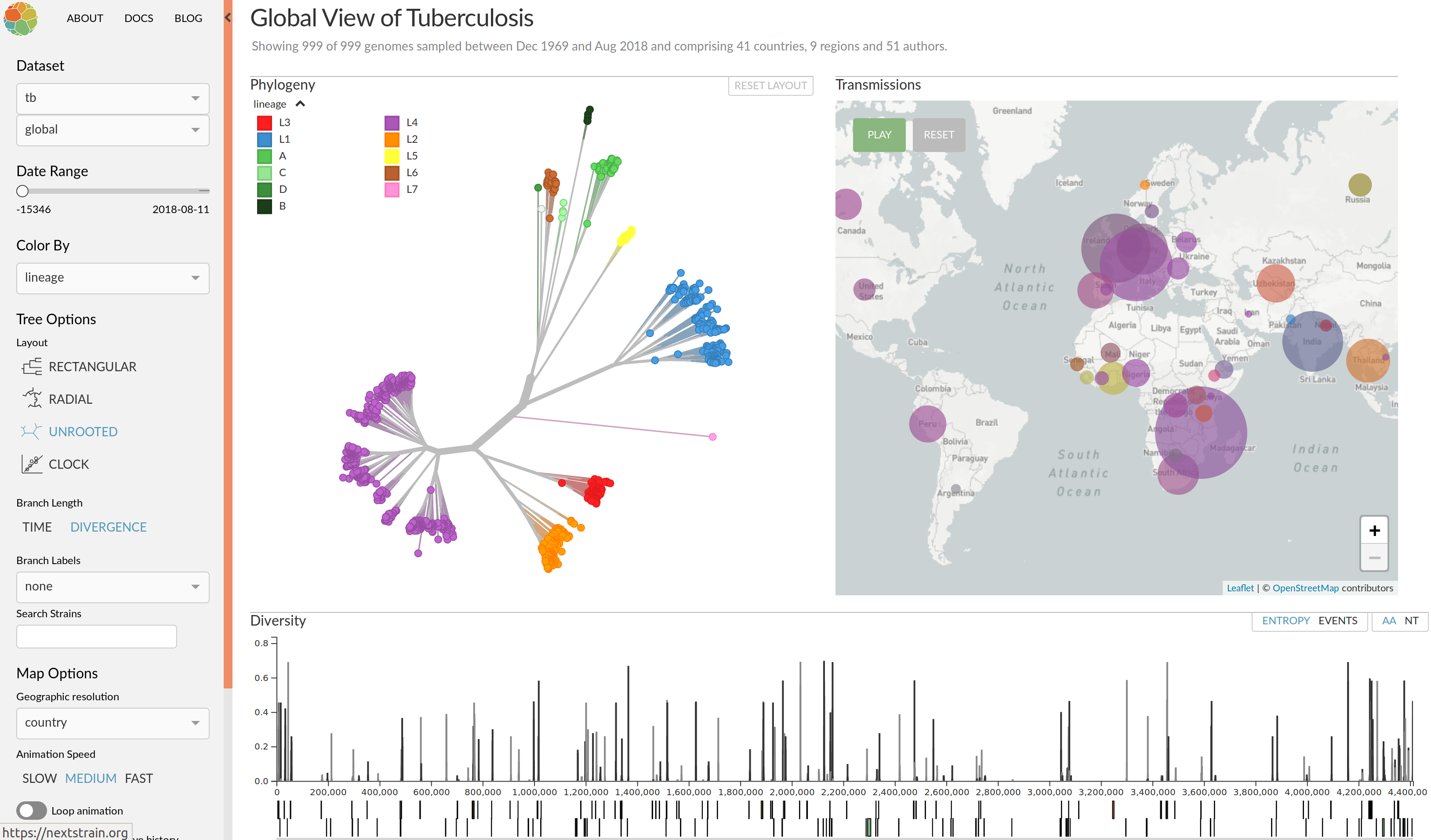
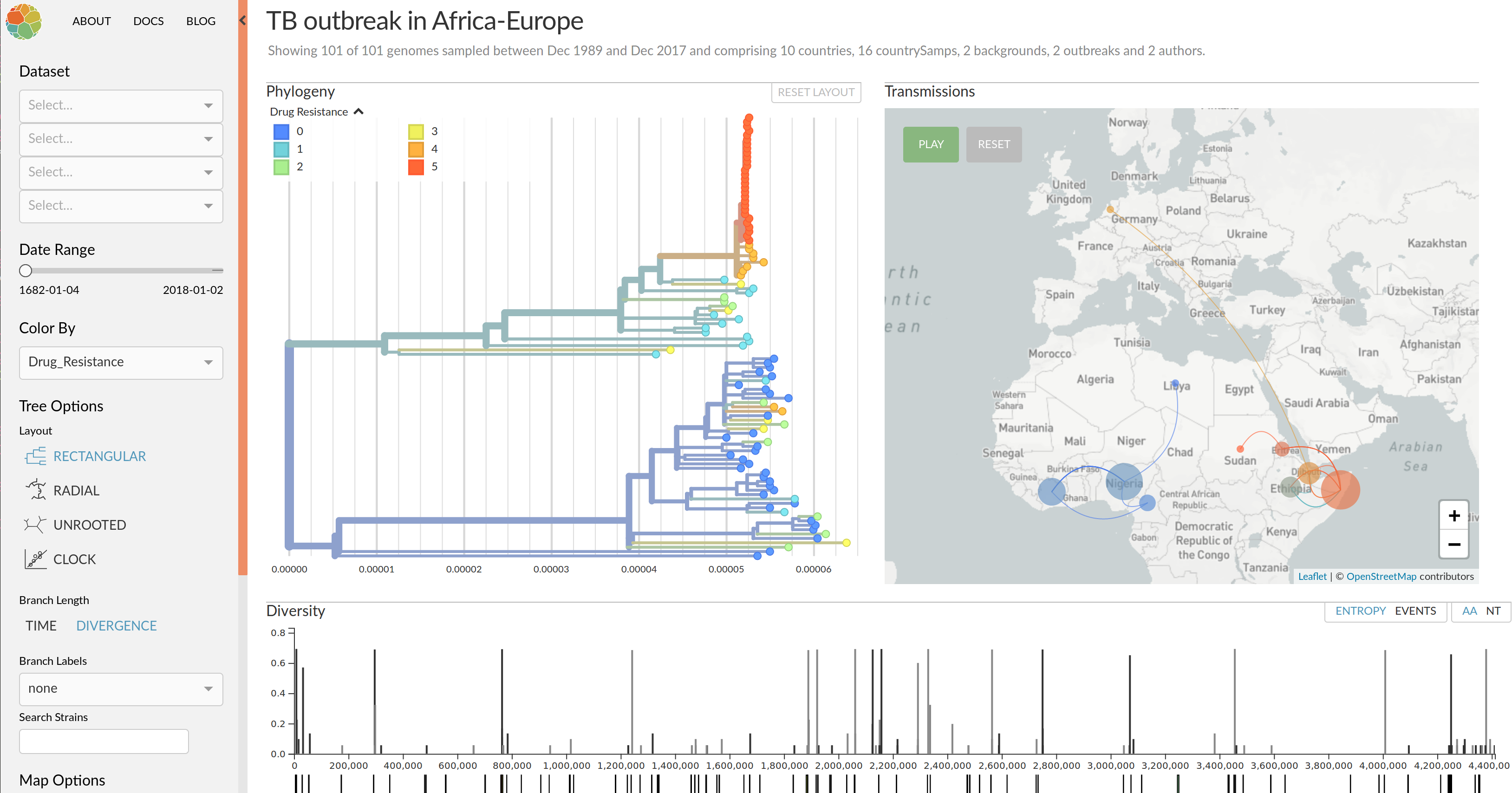
nextflu.org
joint project with Trevor Bedford & his lab
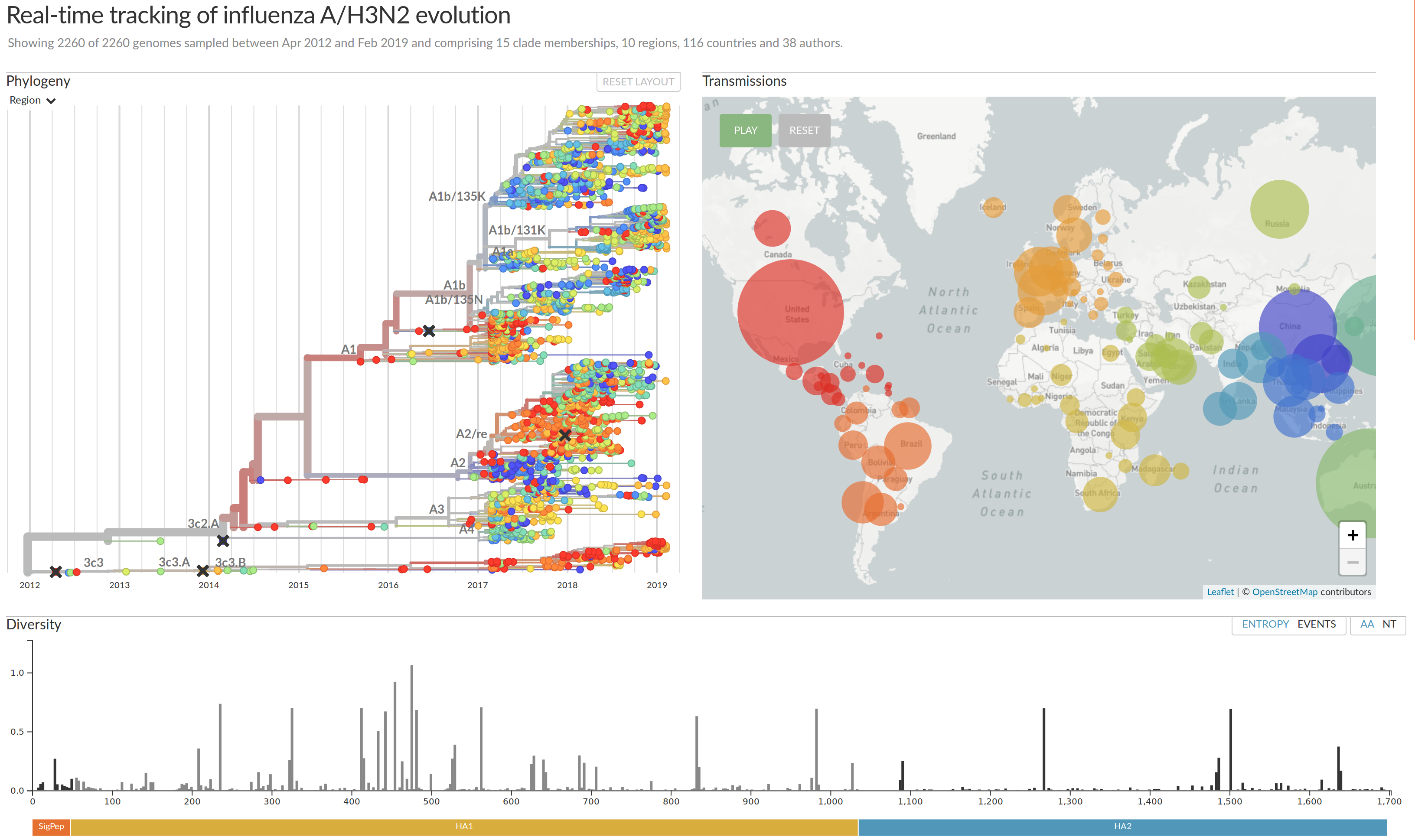
Visualization features of nextstrain
- Regular and time scaled phylogenies
- Mutations are mapped to the tree
- Filtering to time interval, region, country, authors, ...
- Zoom into clades
- Information on specific viruses
- Color by amino acid or nucleotide
- Frequency trajectories of clades and mutations
- Color by antigenic advance, predictive scores, etc
Beyond tracking: can we predict?
Fitness variation in rapidly adapting populations
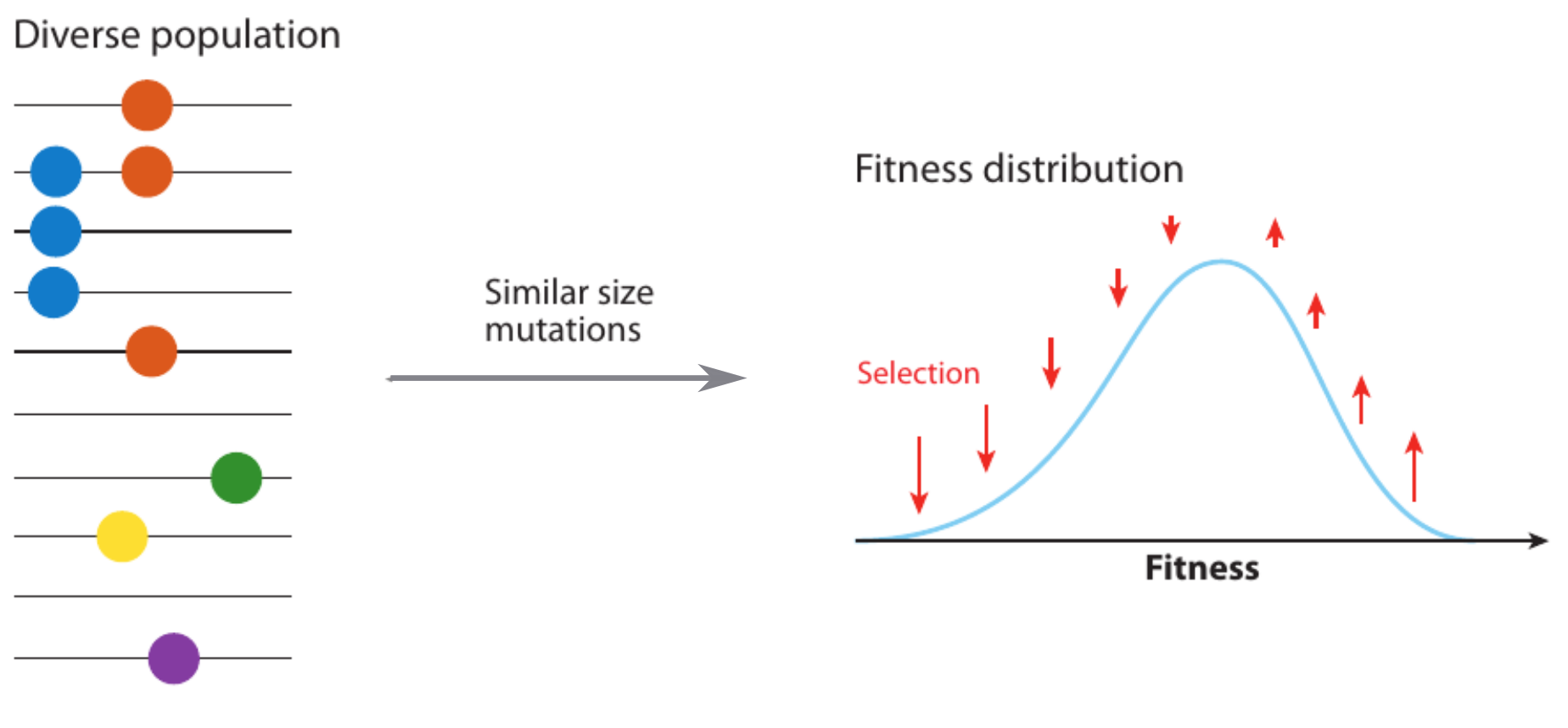
- Speed of adaptation is logarithmic in population size
- Environment (fitness landscape), not mutation supply, determines adaptation
- Different models have universal emerging properties
Predicting evolution
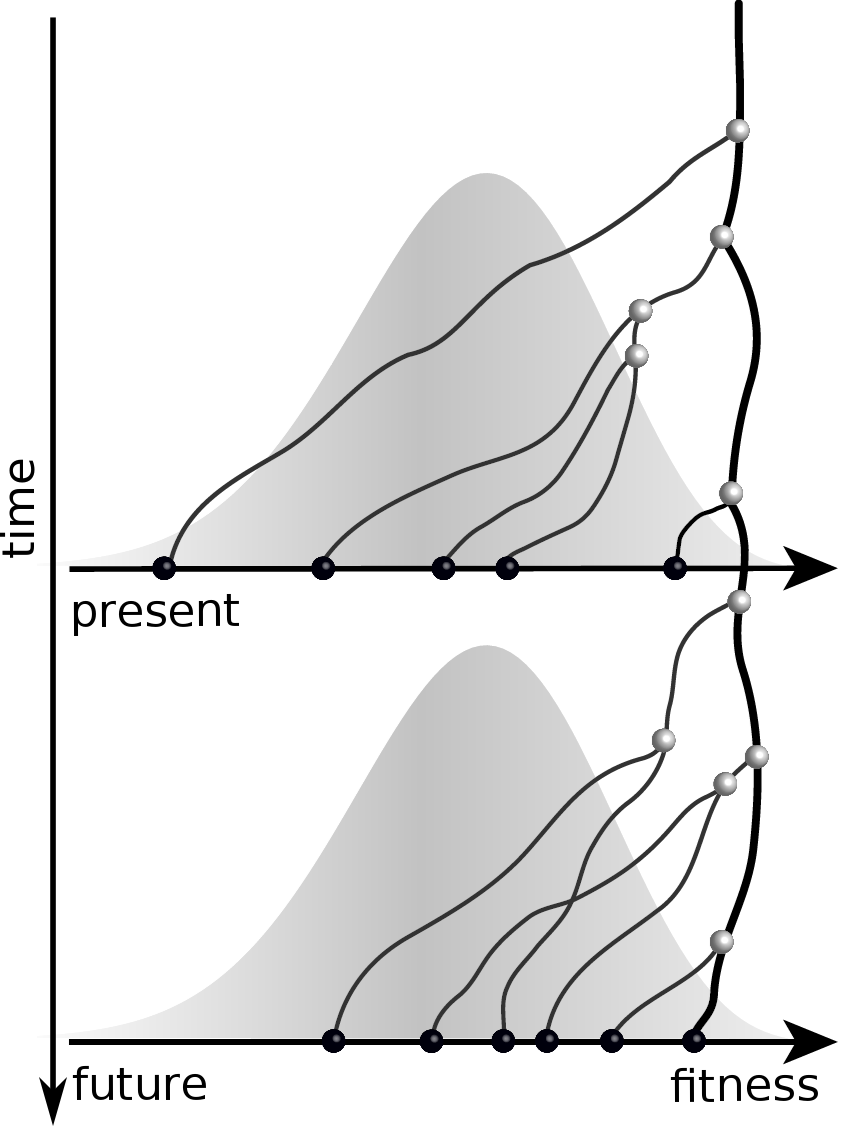
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
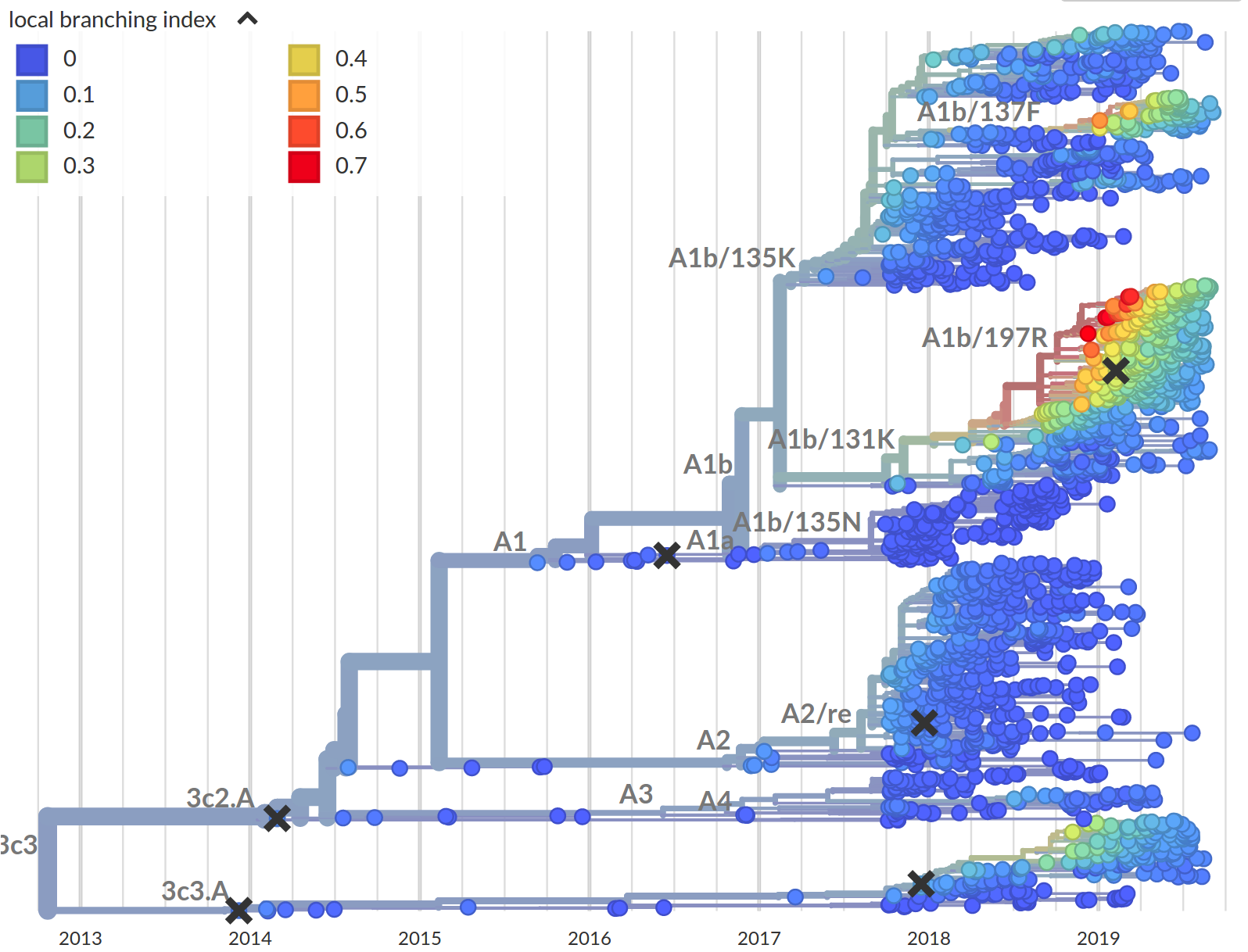
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
Our current prediction...

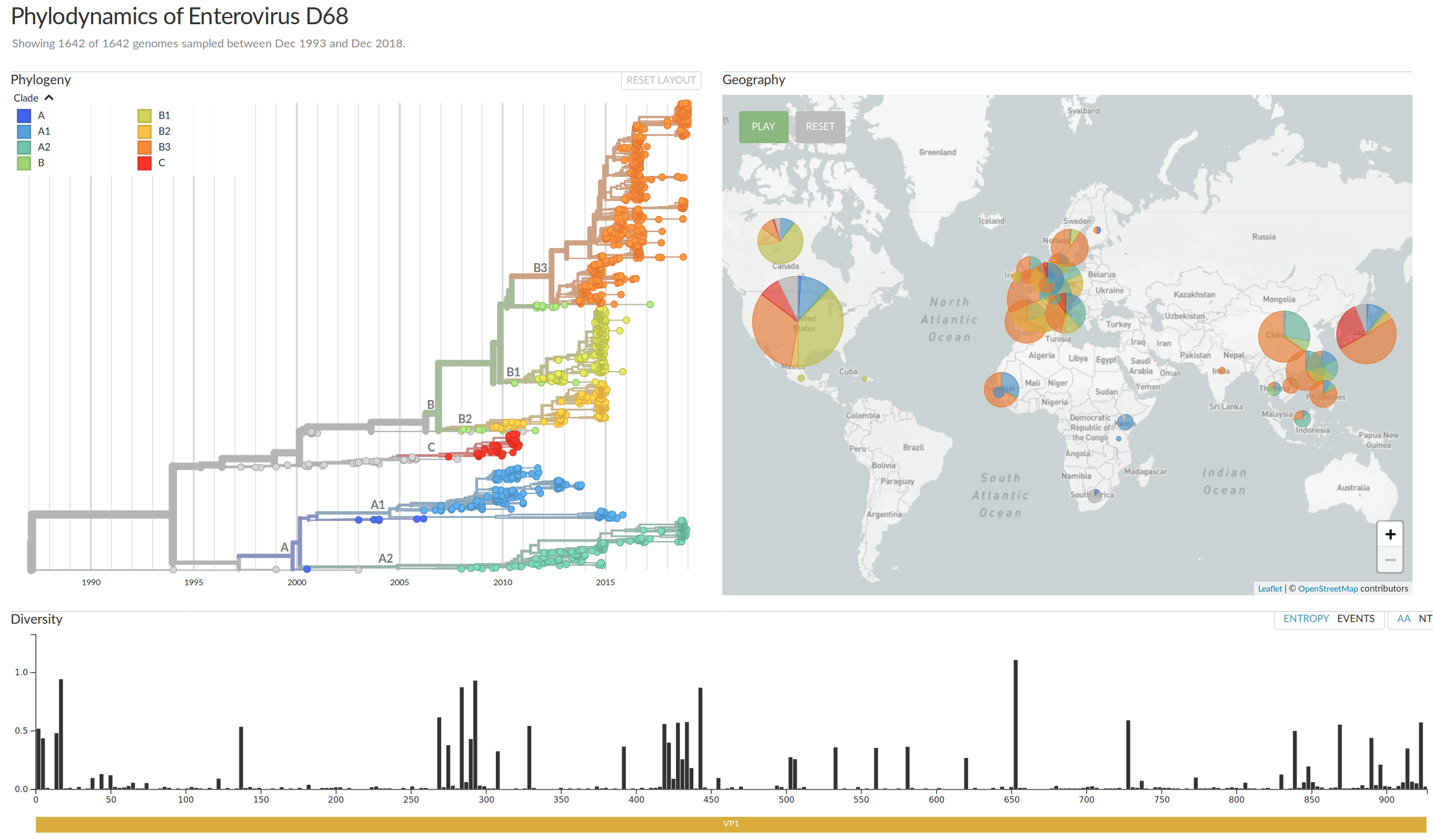
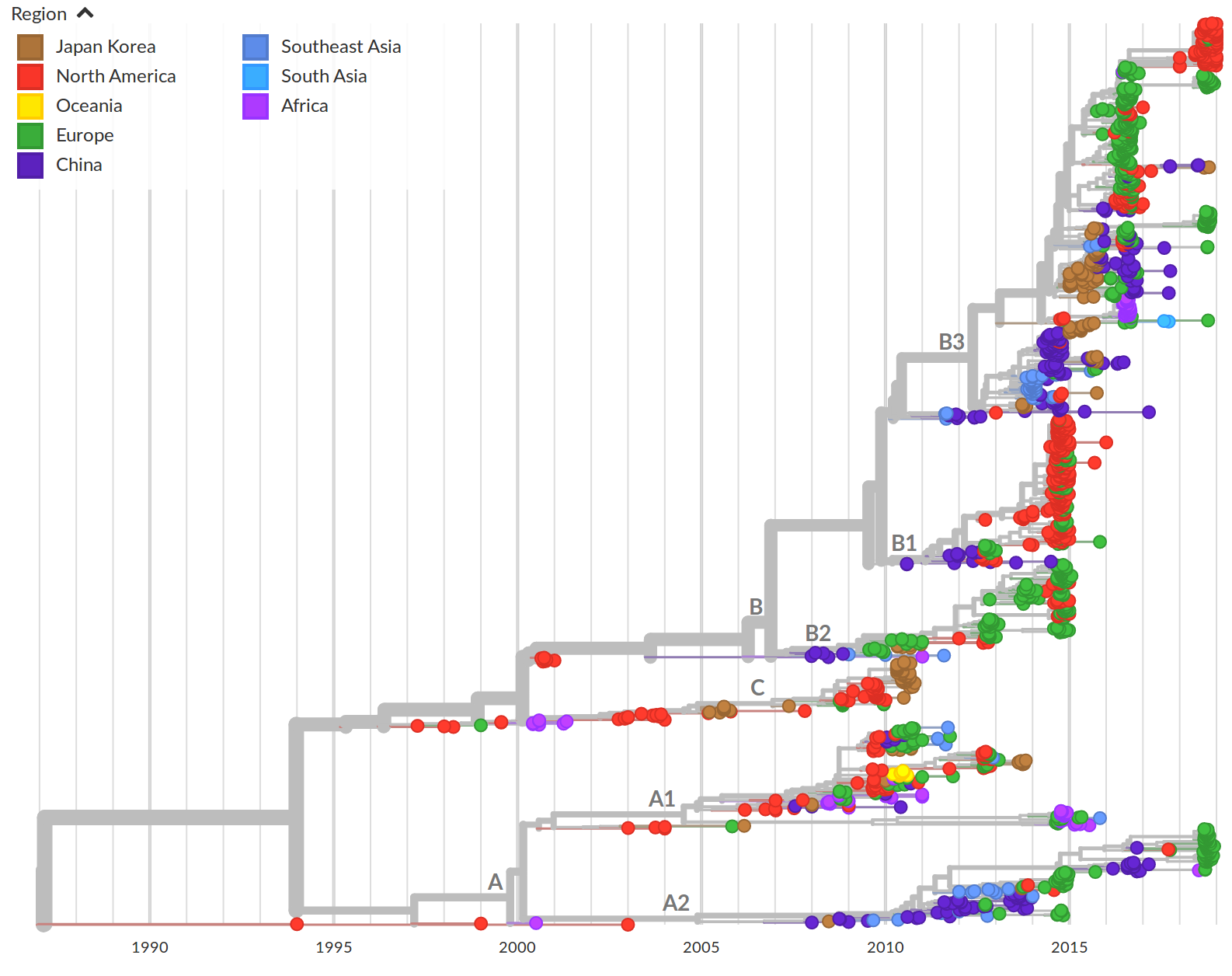
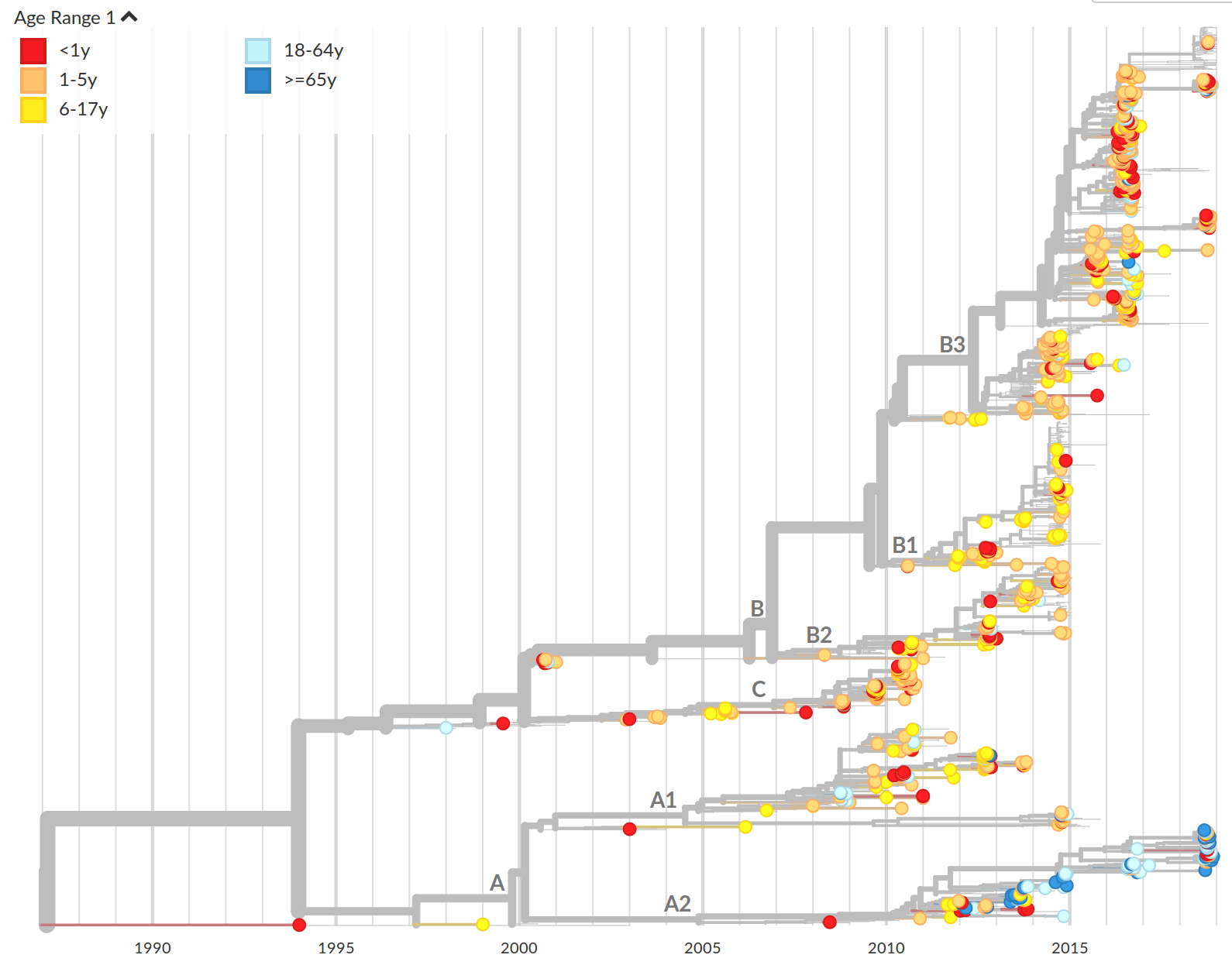
Geographic and demographic distribution EV-D68
Acknowledgments



- Trevor Bedford
- Pavel Sagulenko
- James Hadfield
- Emma Hodcroft
- Tom Sibley
- and others

Influenza and Theory acknowledgments



- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
Acknowledgments -- Enterovirus
- Robert Dyrdak
- Jan Albert
- Lina Thebo
- Emma Hodcroft
- Bert Niesters (Groningen)
- Randy Poelman (Groningen)
- Elke Wollants (Leuven)
- Adrian Egli (Basel)
- Andrés Antón Pagarolas (Barcelona)