Tracking SARS-CoV-2 using real-time phylogenetics with Nextstrain
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/202004_TAGC.html
I won't talk specifically about work by myself or by my group
Instead:
Overview of a collective effort from the Nextstrain perspective
Acknowledgements
Trevor Bedford and his lab -- terrific collaboration since 2014

James Hadfield, Emma Hodcroft and Tom Sibley have led the recent development
Data we analyze are contributed by scientists from all over the world
Data are shared and curated by GISAID

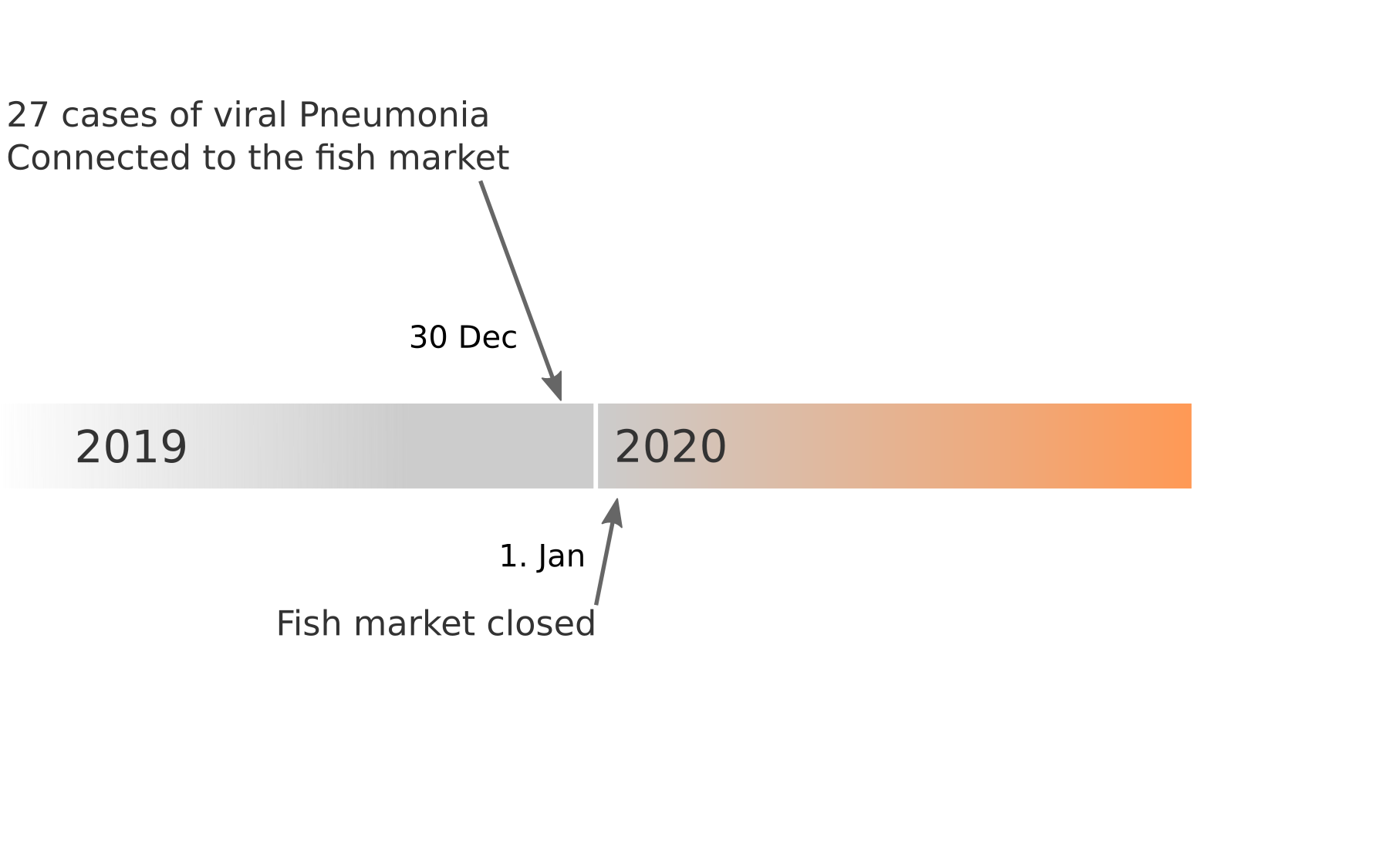 Data summarized by Ian MacKay
Data summarized by Ian MacKay
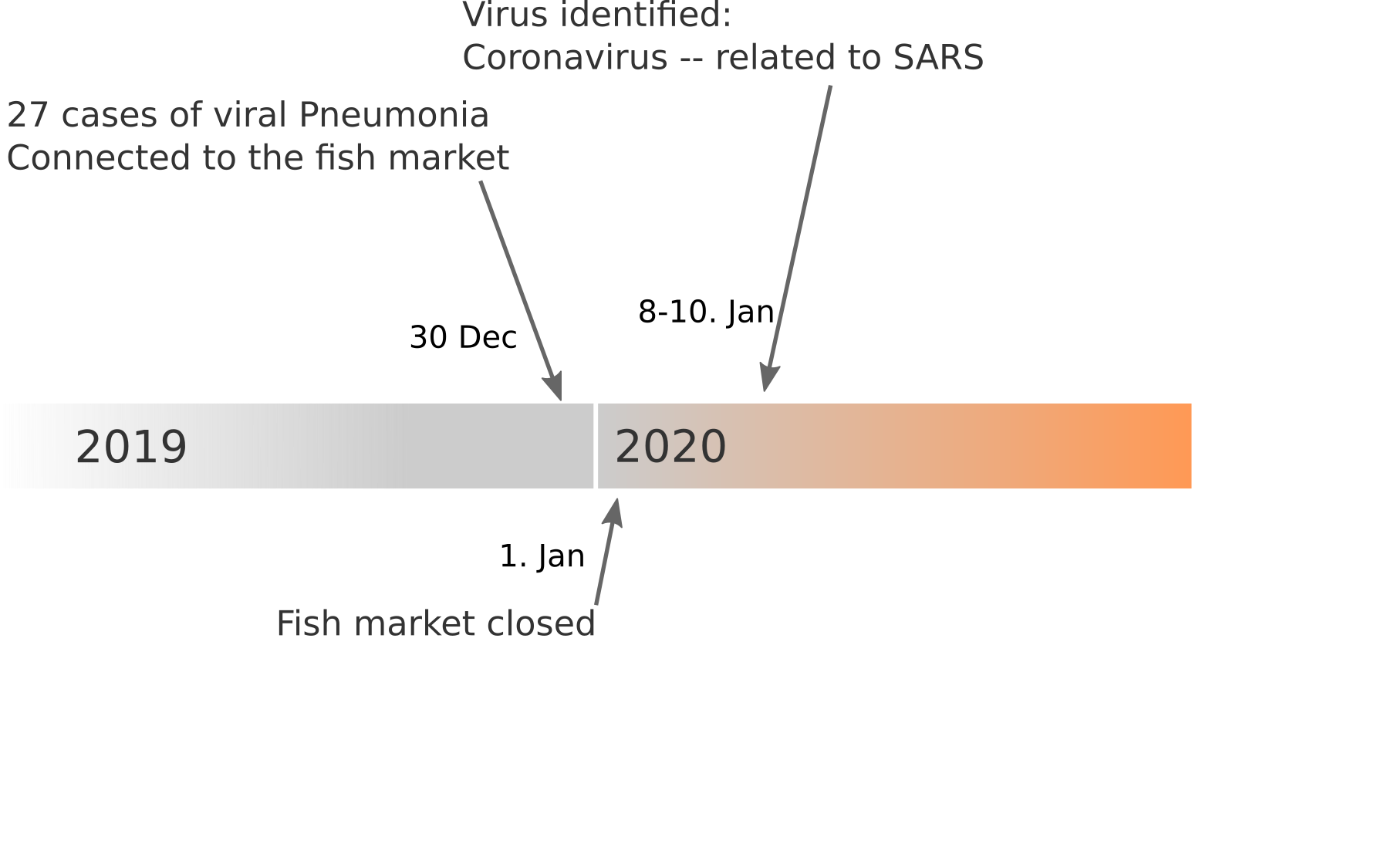 Data summarized by Ian MacKay
Data summarized by Ian MacKay
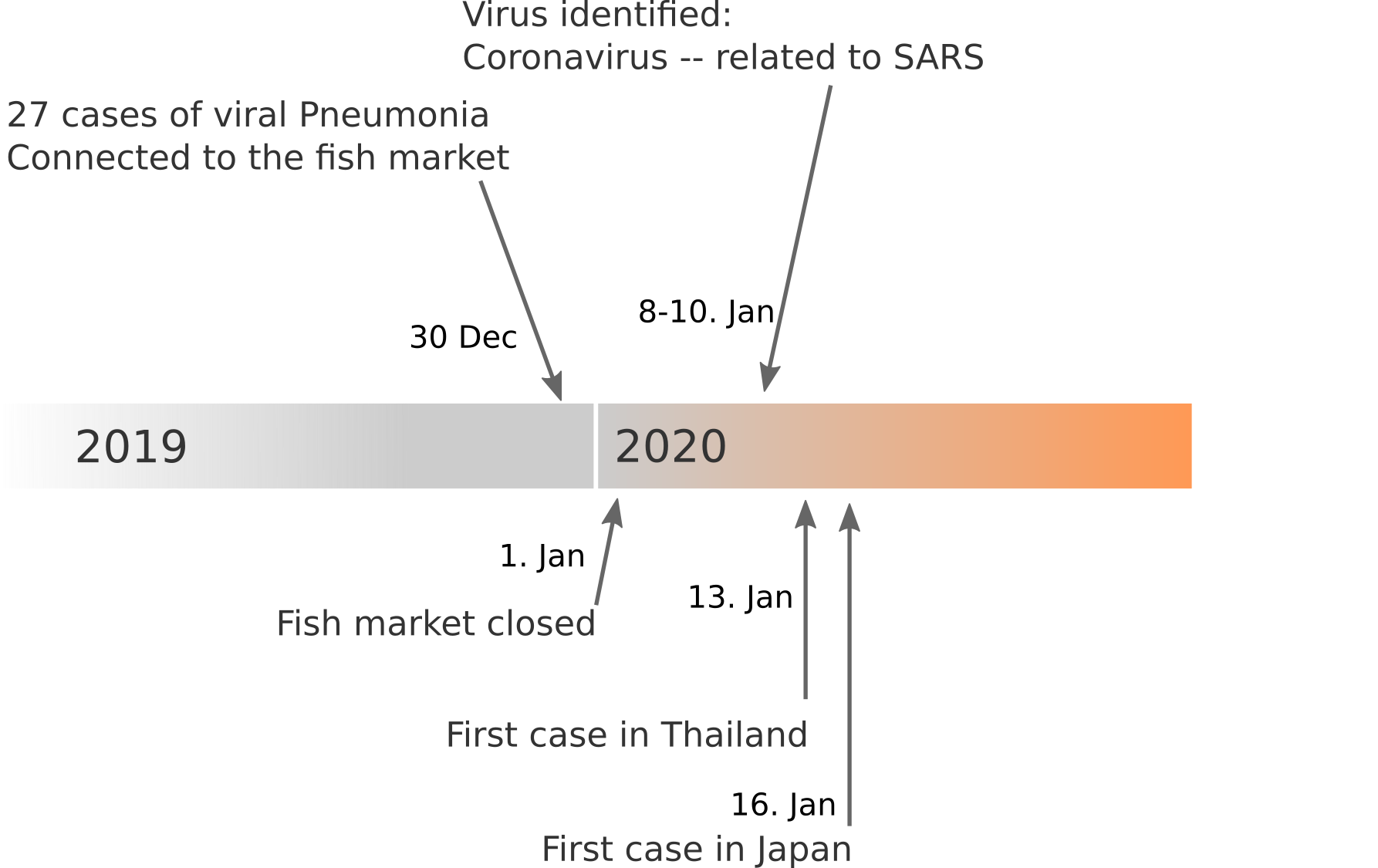 Data summarized by Ian MacKay
Data summarized by Ian MacKay
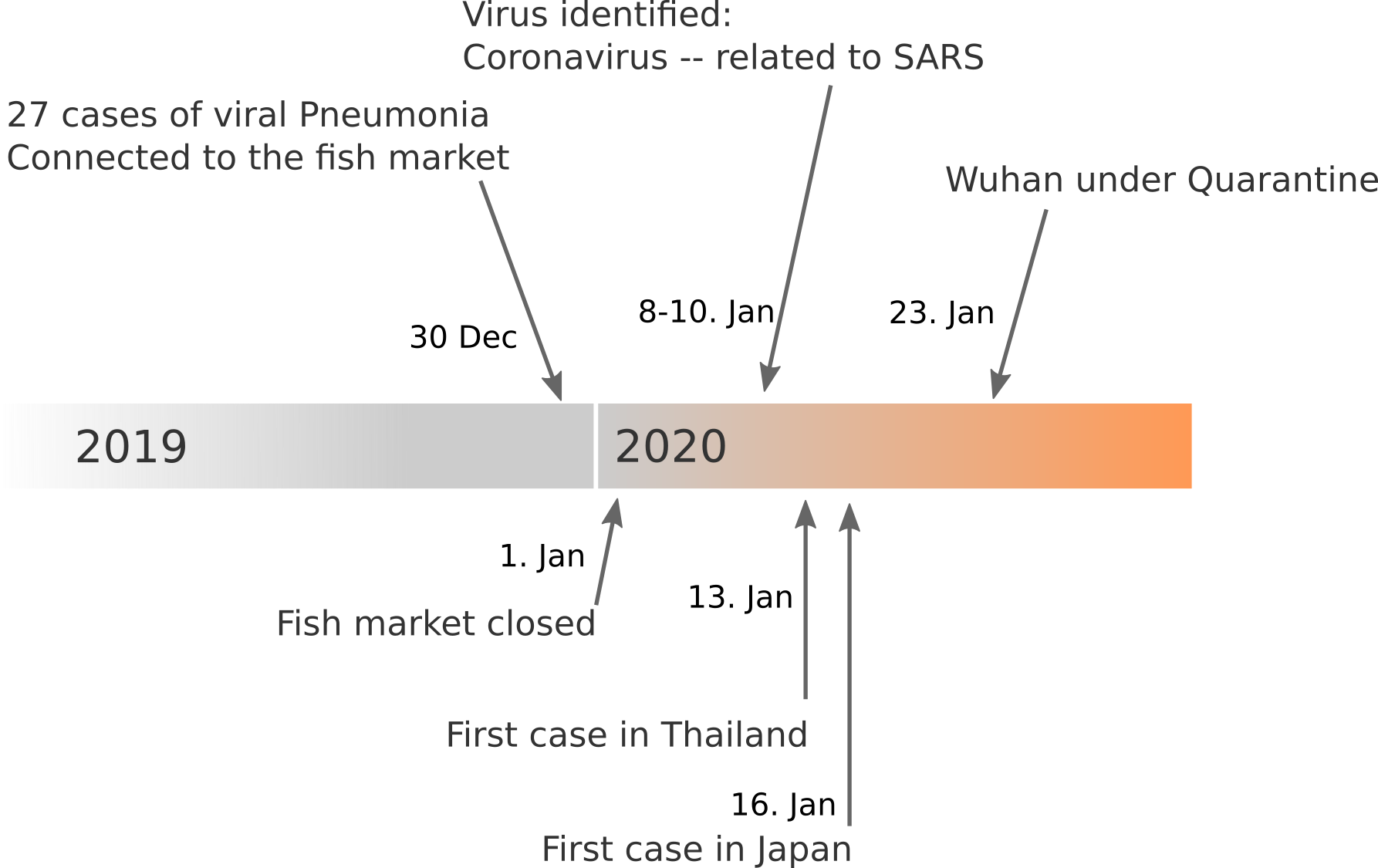 Data summarized by Ian MacKay
Data summarized by Ian MacKay
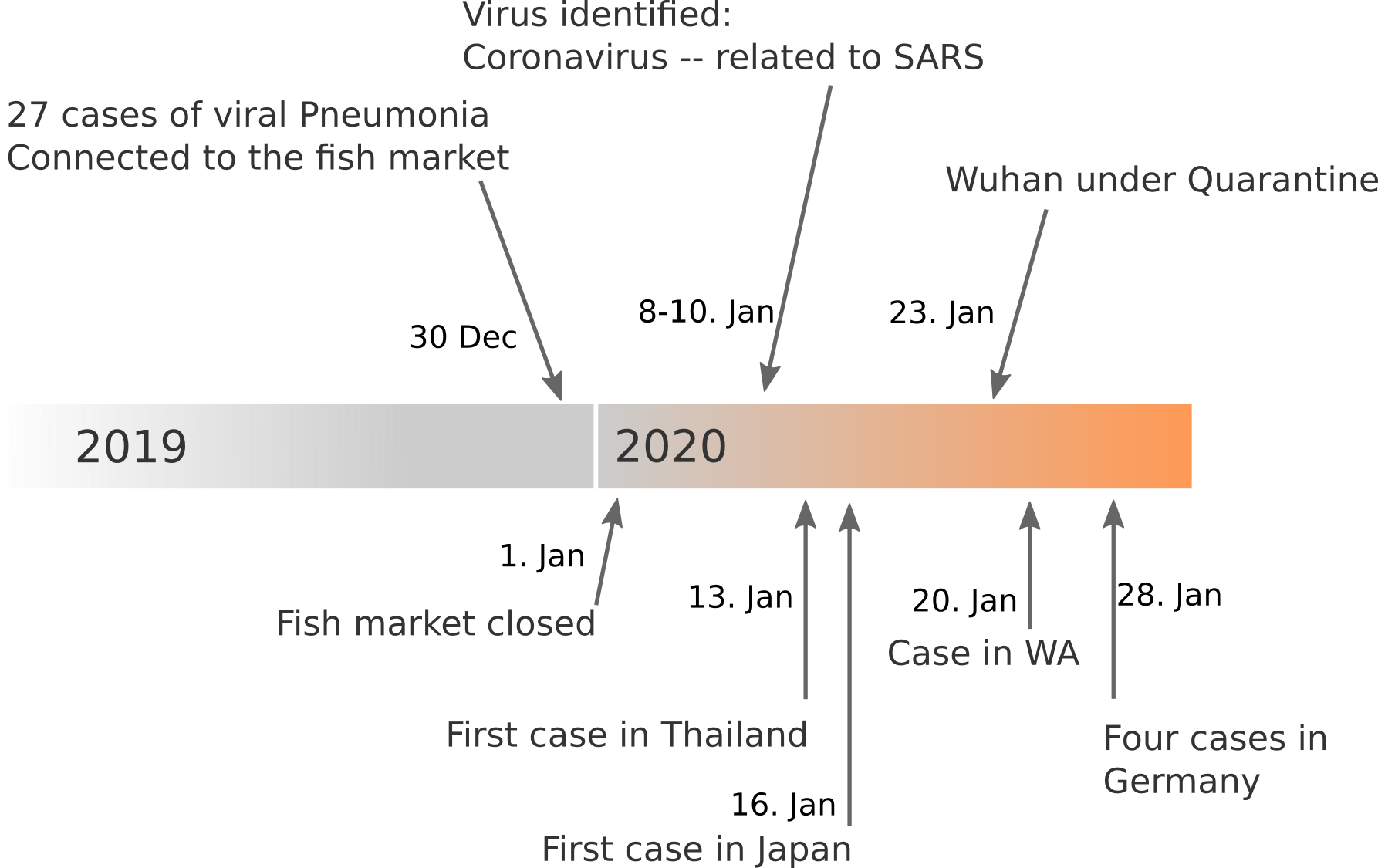 Data summarized by Ian MacKay
Data summarized by Ian MacKay
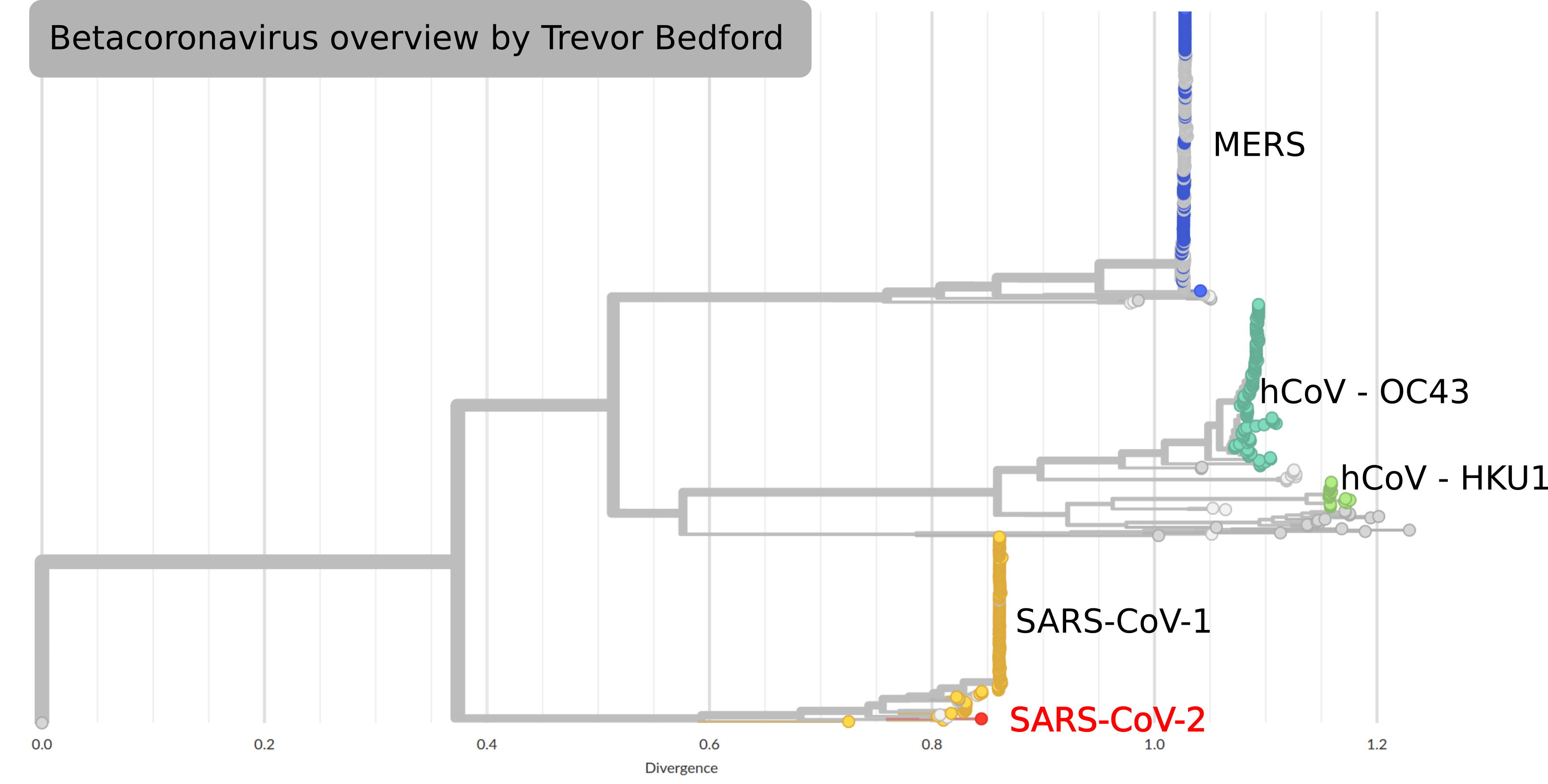 by Trevor Bedford
by Trevor Bedford
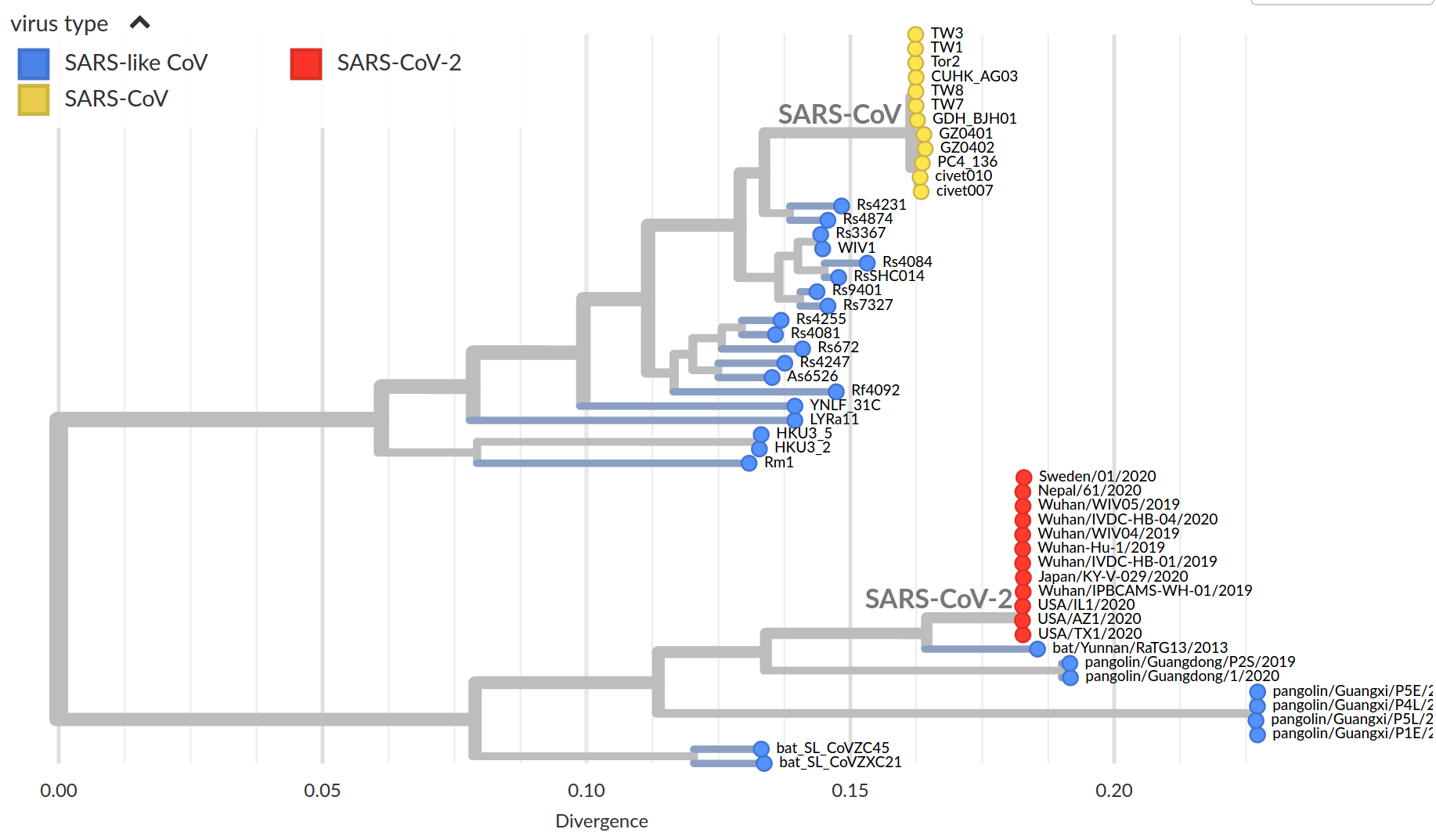 by Trevor Bedford
by Trevor Bedford
Tracking diversity and spread of SARS-CoV-2 in nextstrain
The SARS-CoV-2 genome
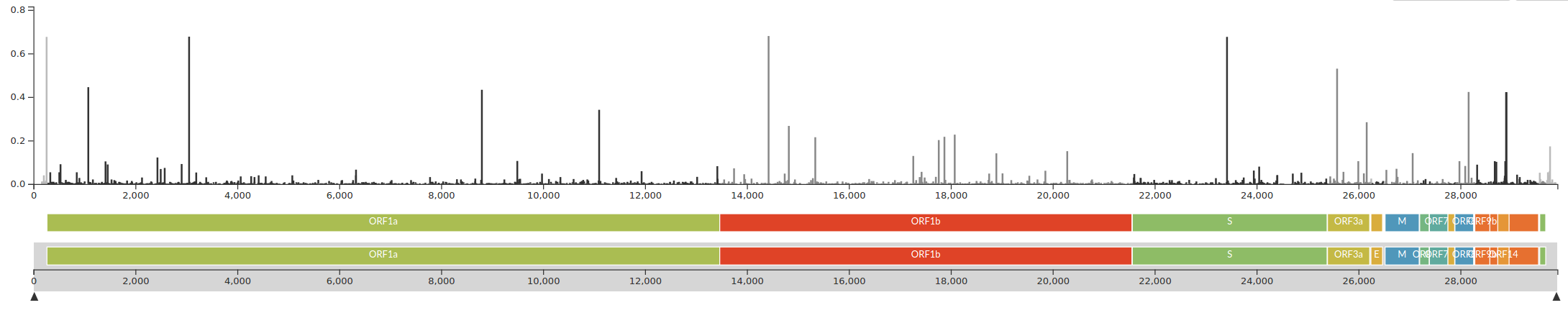
- 29k linear (+)ssRNA genome -- one of largest RNA virus genomes
- the first 2/3 code for the replication machinery
Available data on Jan 26
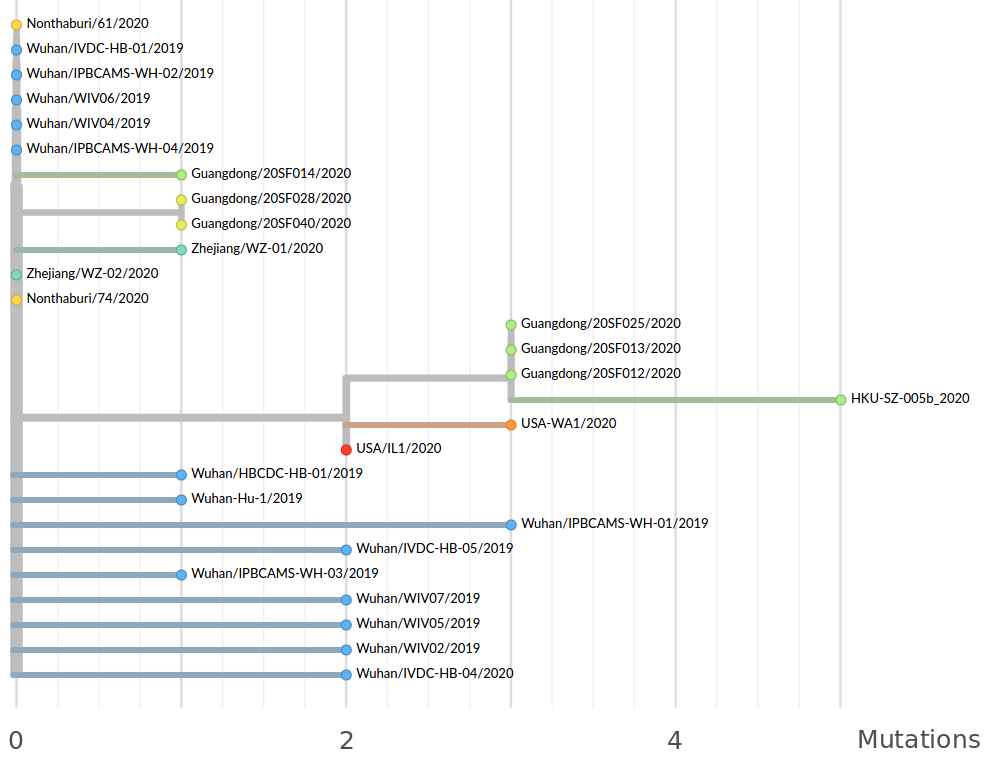
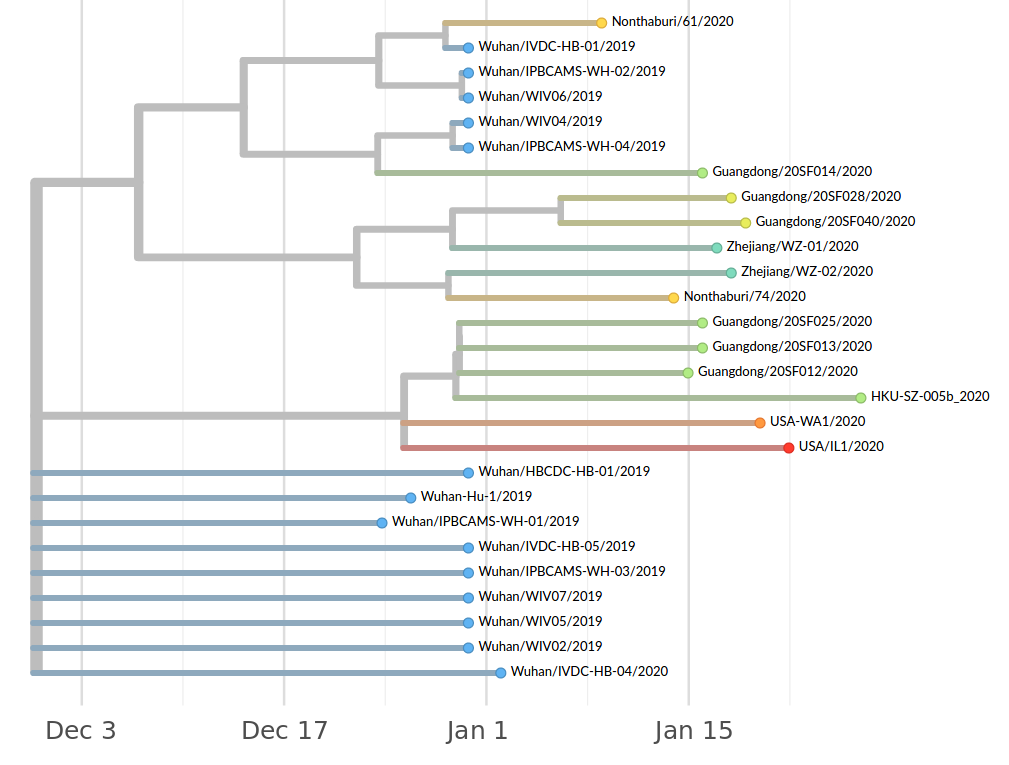
Early genomes differed by only a few mutations, suggesting very recent emergence
→ the closest to "real-time" we have experienced so far...
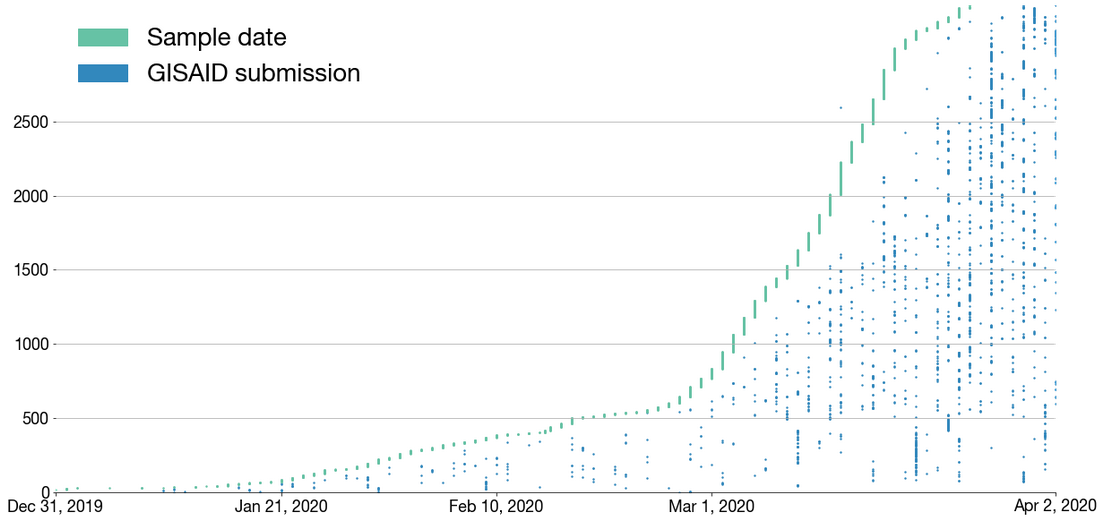
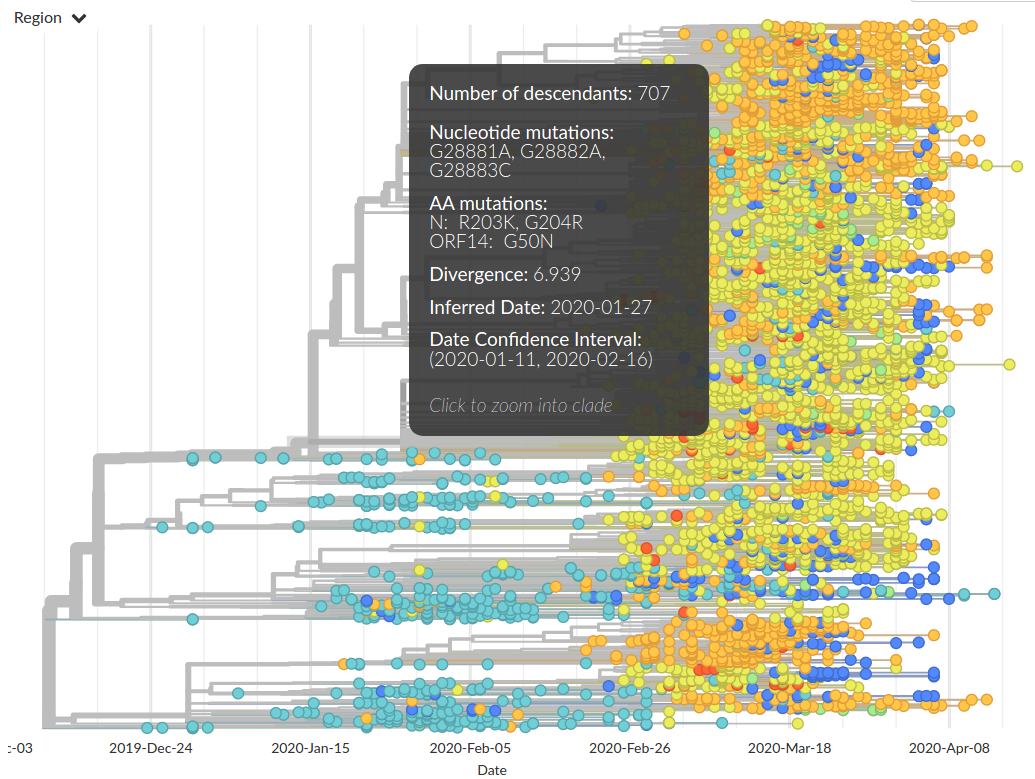
Figure by James HadfieldAvailable data on April 22
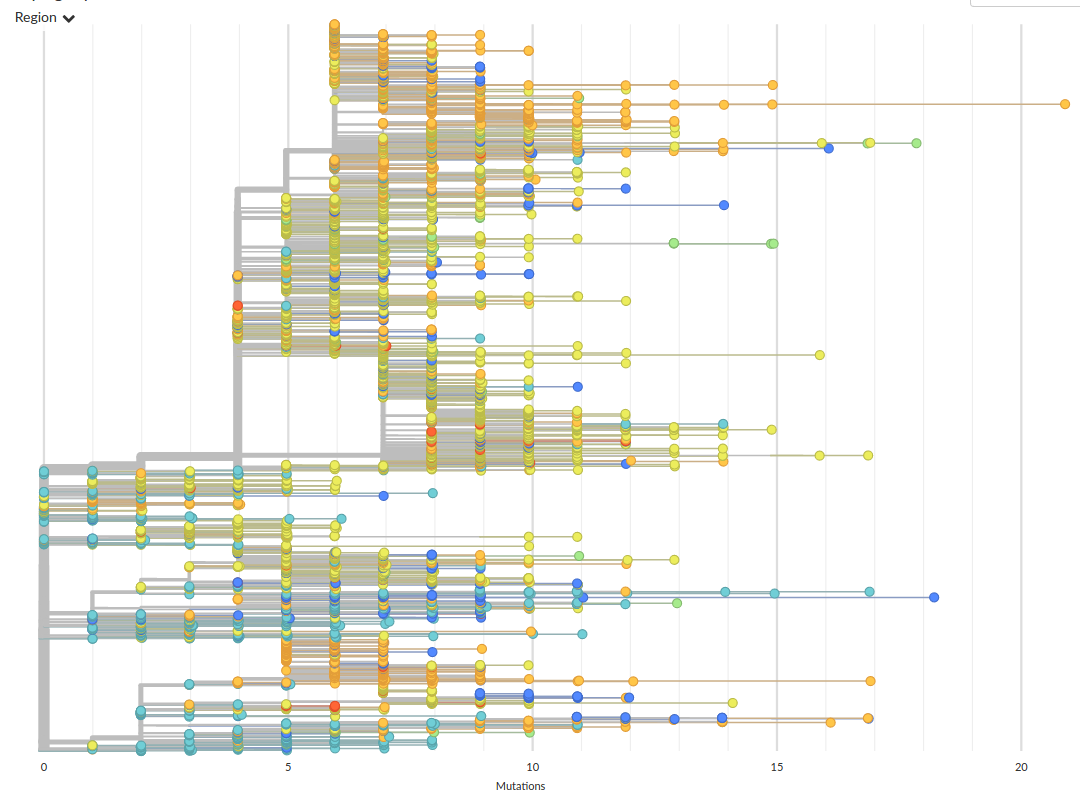
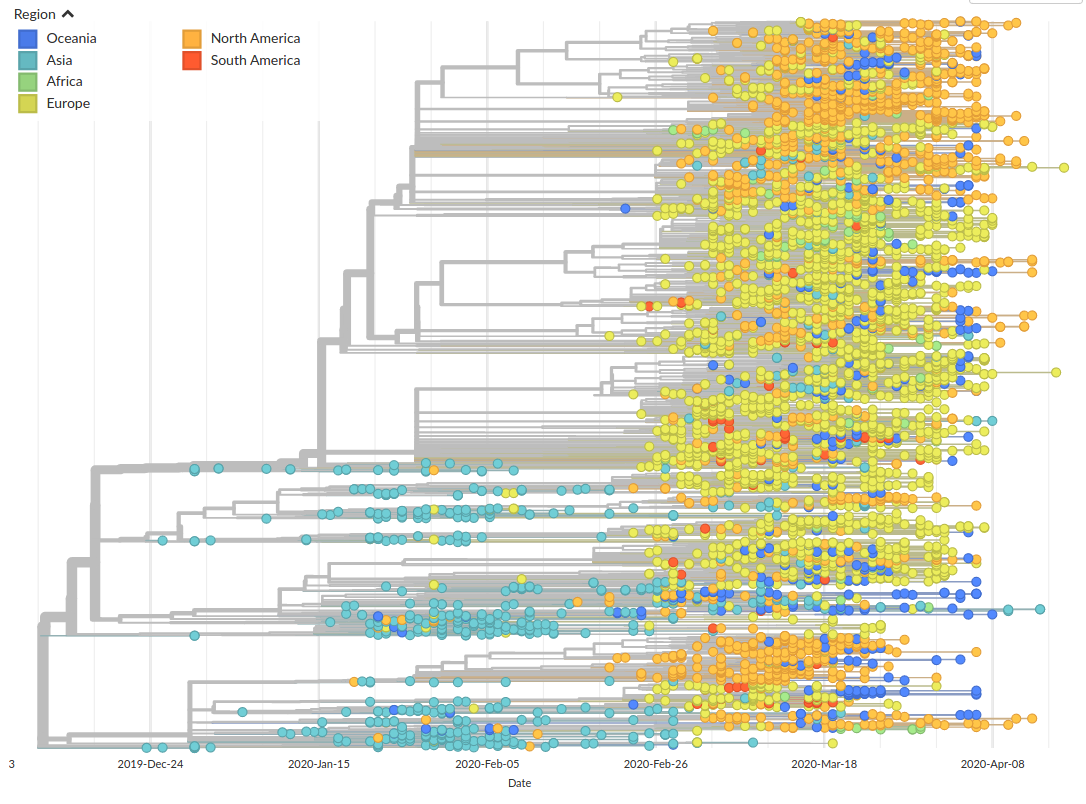
Subset of available data on April 22
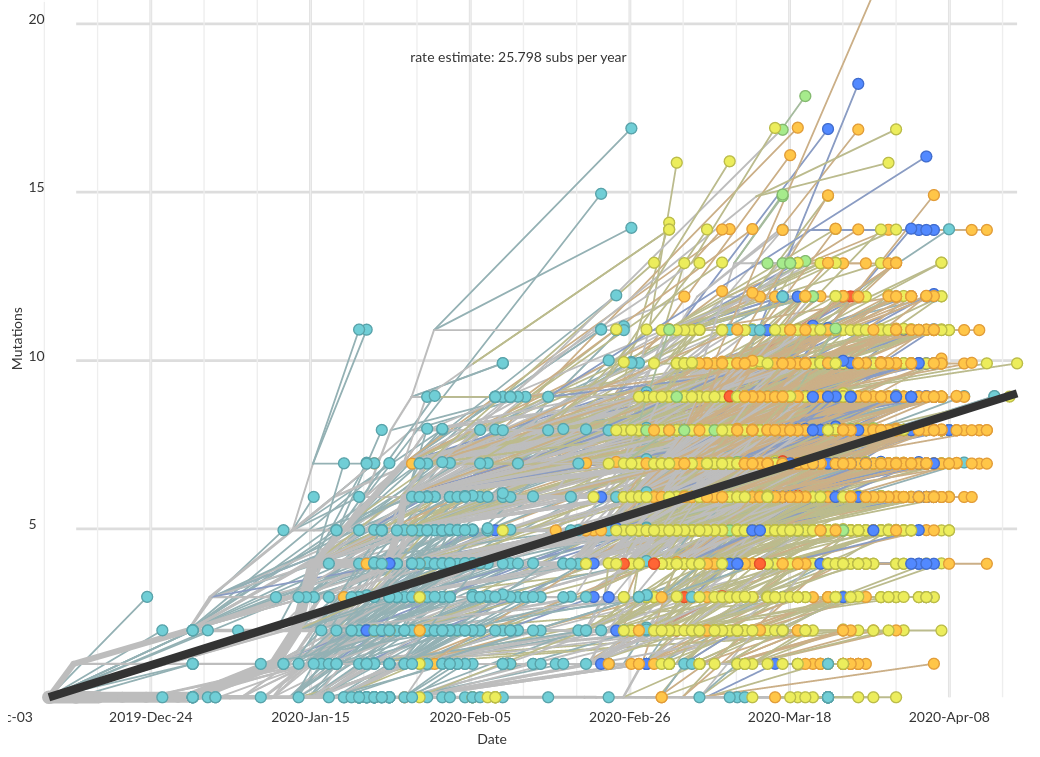
About two mutations per month, but mutations are often clustered and overdispersed
Tracing the origins of samples from Iceland
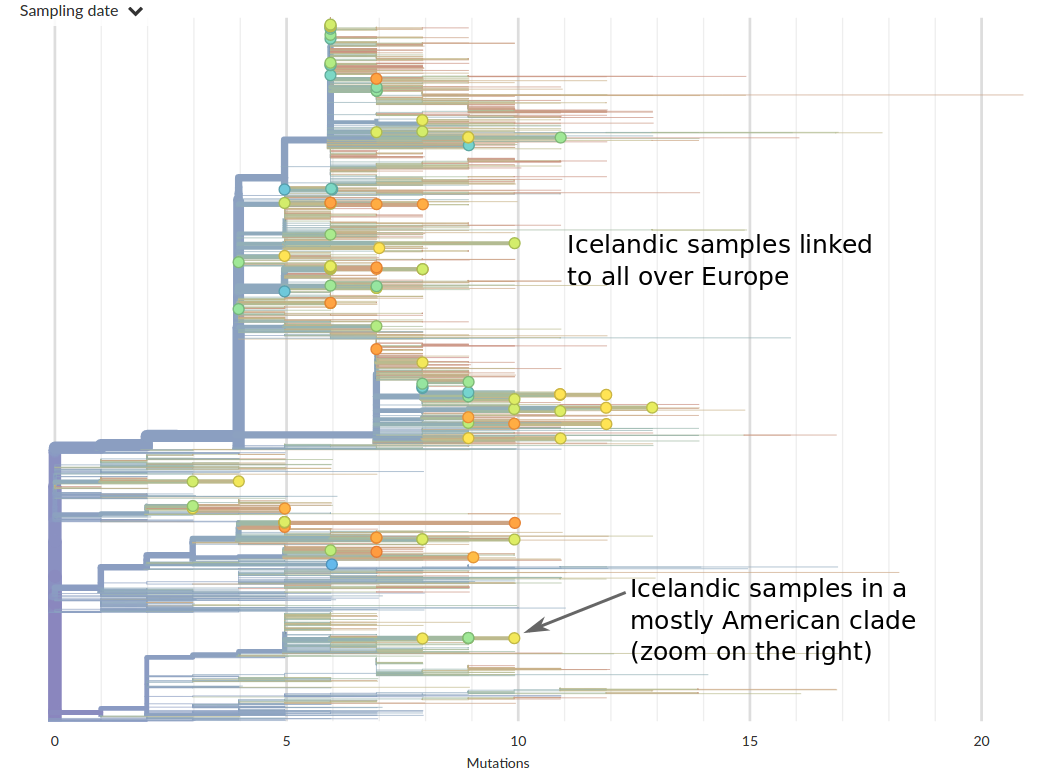
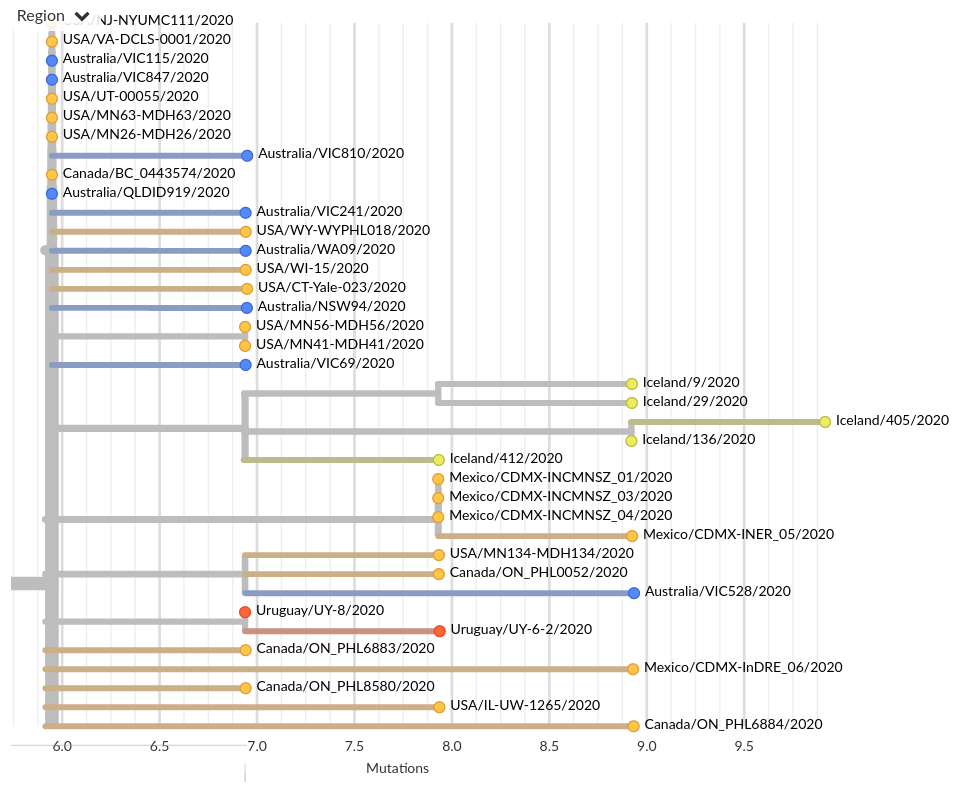
SARS-CoV-2 is remarkably mixed geographically -- genomes connect outbreaks in different places
Fighting misinformation
Colorful trees are easily misinterpreted...
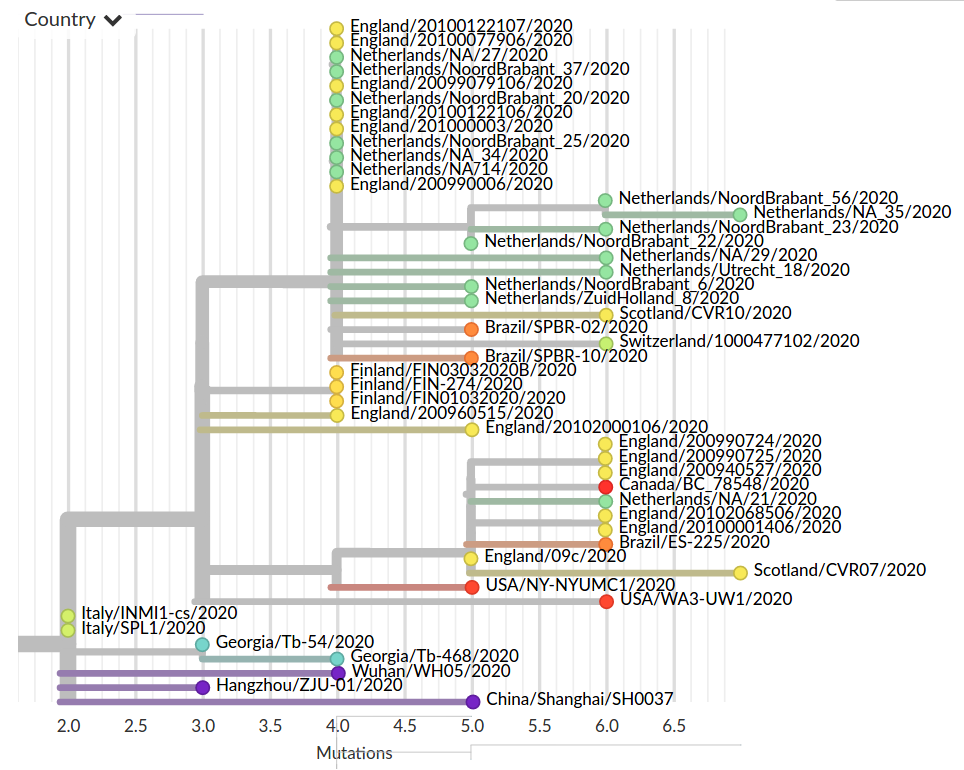
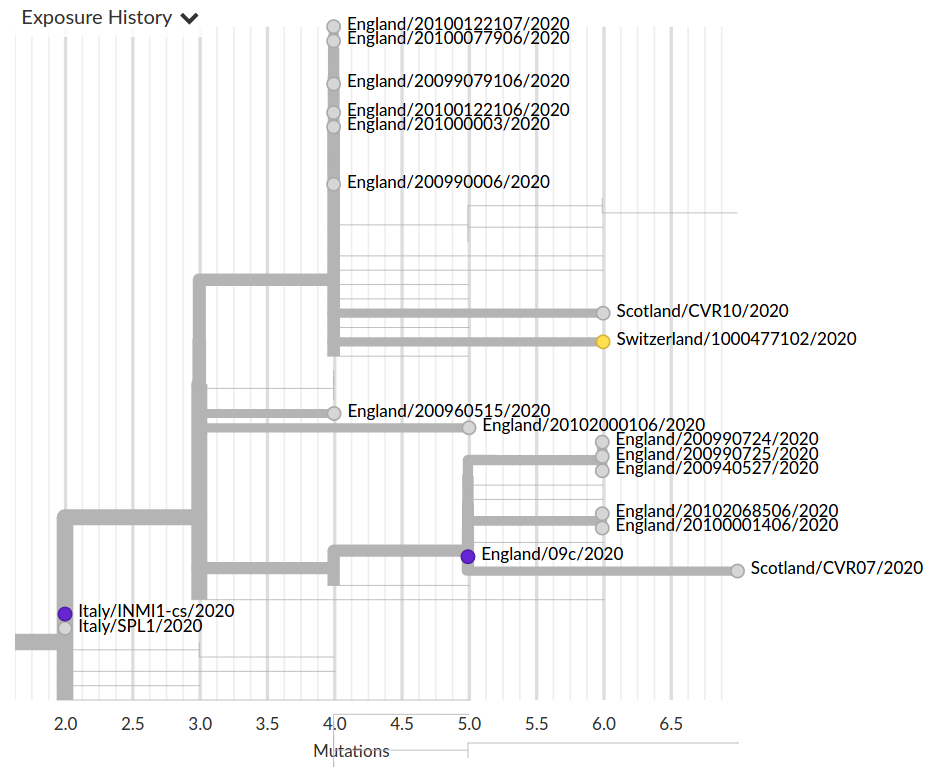
Low genetic diversity combined with very biased sampling → no directionality can be inferred
Nextstrain situation reports
Sidney Bell, Cassia Wagner, Emma Hodcroft, James Hadfield, Nicola Müller and others
Acknowledgements
Trevor Bedford and his lab -- terrific collaborations since 2014