Real-time tracking of SARS-CoV-2 spread and evolution
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/202007_ISMB.html
Acknowledgments
Trevor Bedford and his lab -- terrific collaboration since 2014

especially James Hadfield, Emma Hodcroft, Ivan Aksamentov, Moira Zuber, and Tom Sibley
Data we analyze are contributed by scientists from all over the world
Data are shared and curated by GISAID

 BBC
BBC
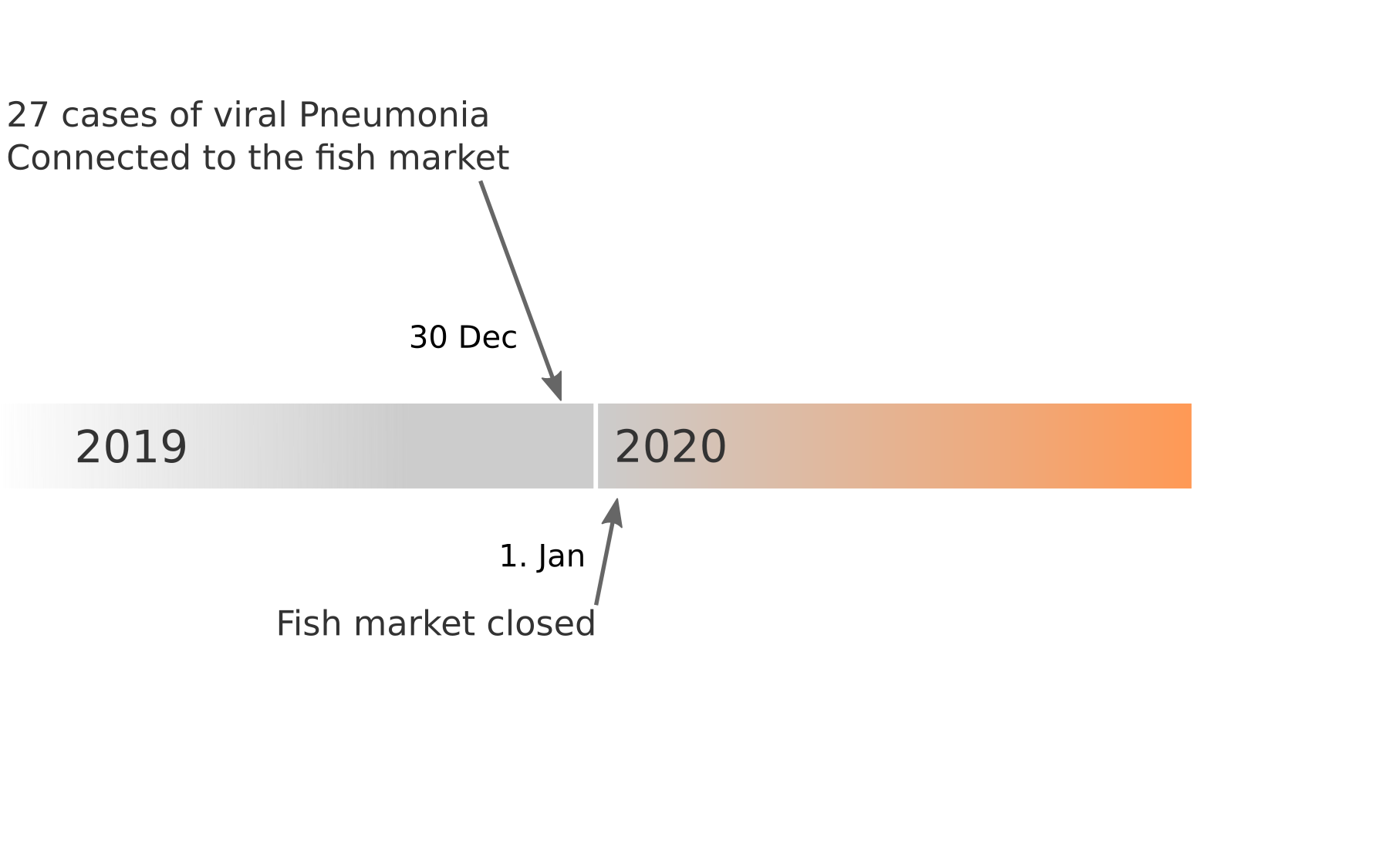 Data summarized by Ian MacKay
Data summarized by Ian MacKay
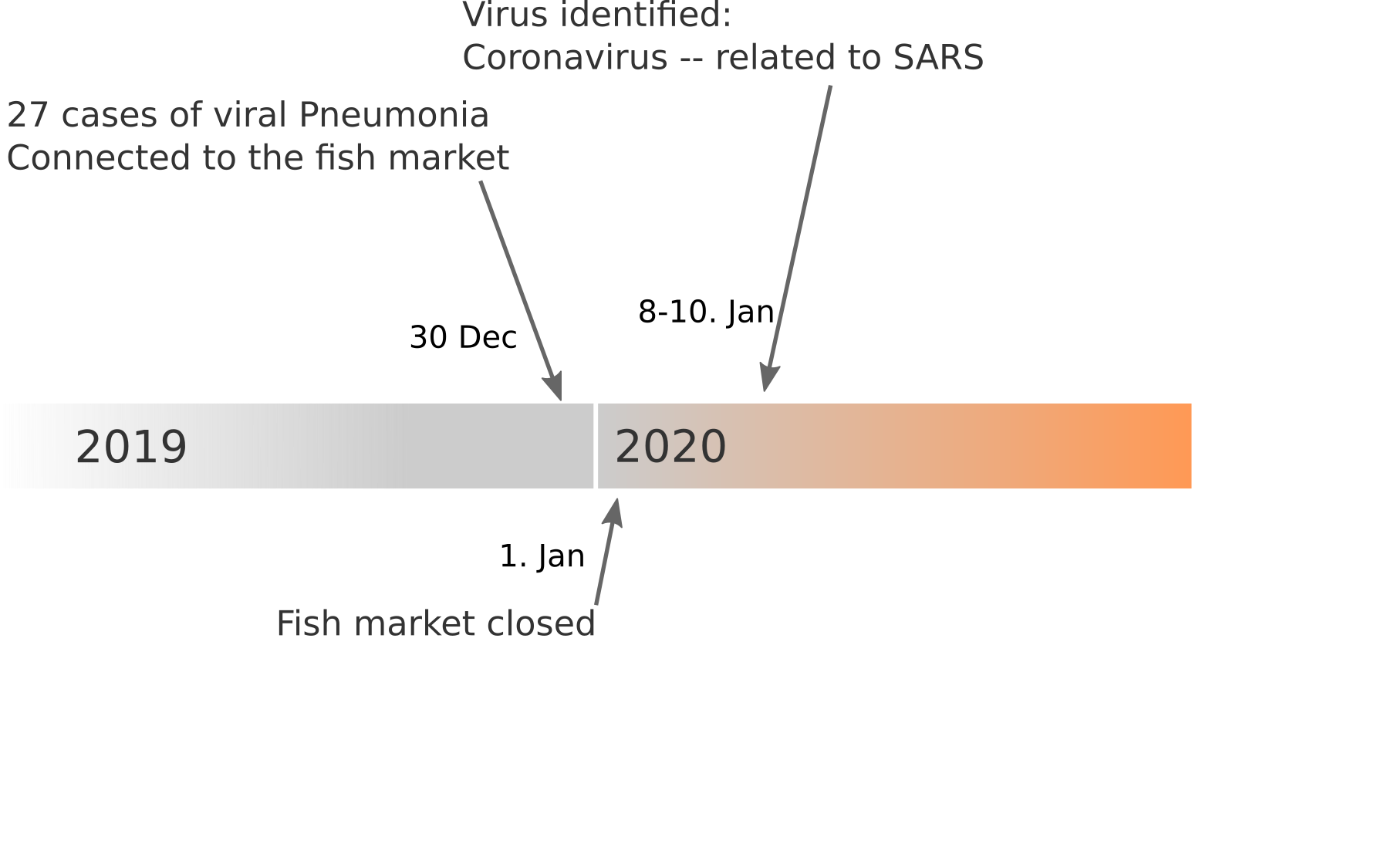 Data summarized by Ian MacKay
Data summarized by Ian MacKay
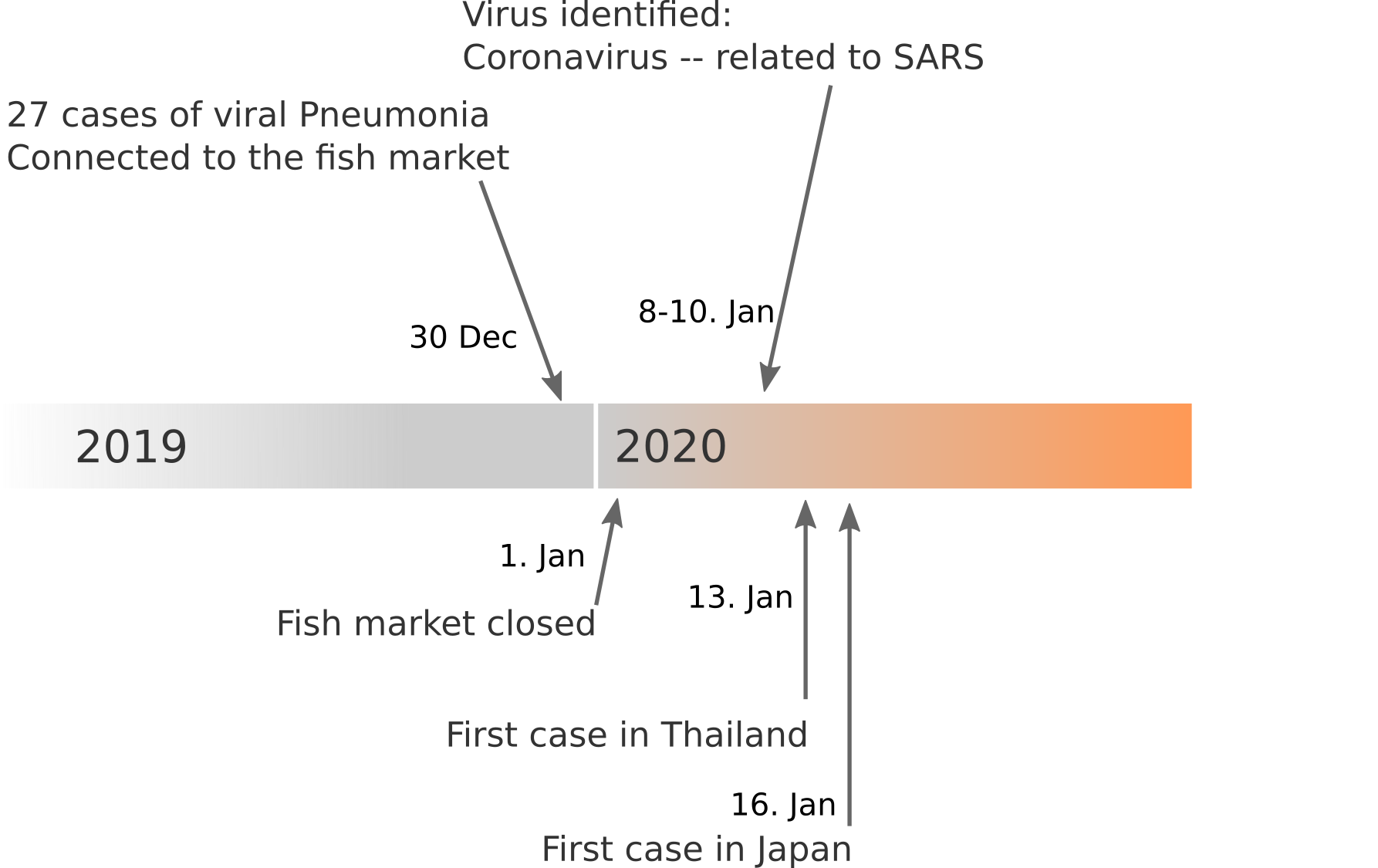 Data summarized by Ian MacKay
Data summarized by Ian MacKay
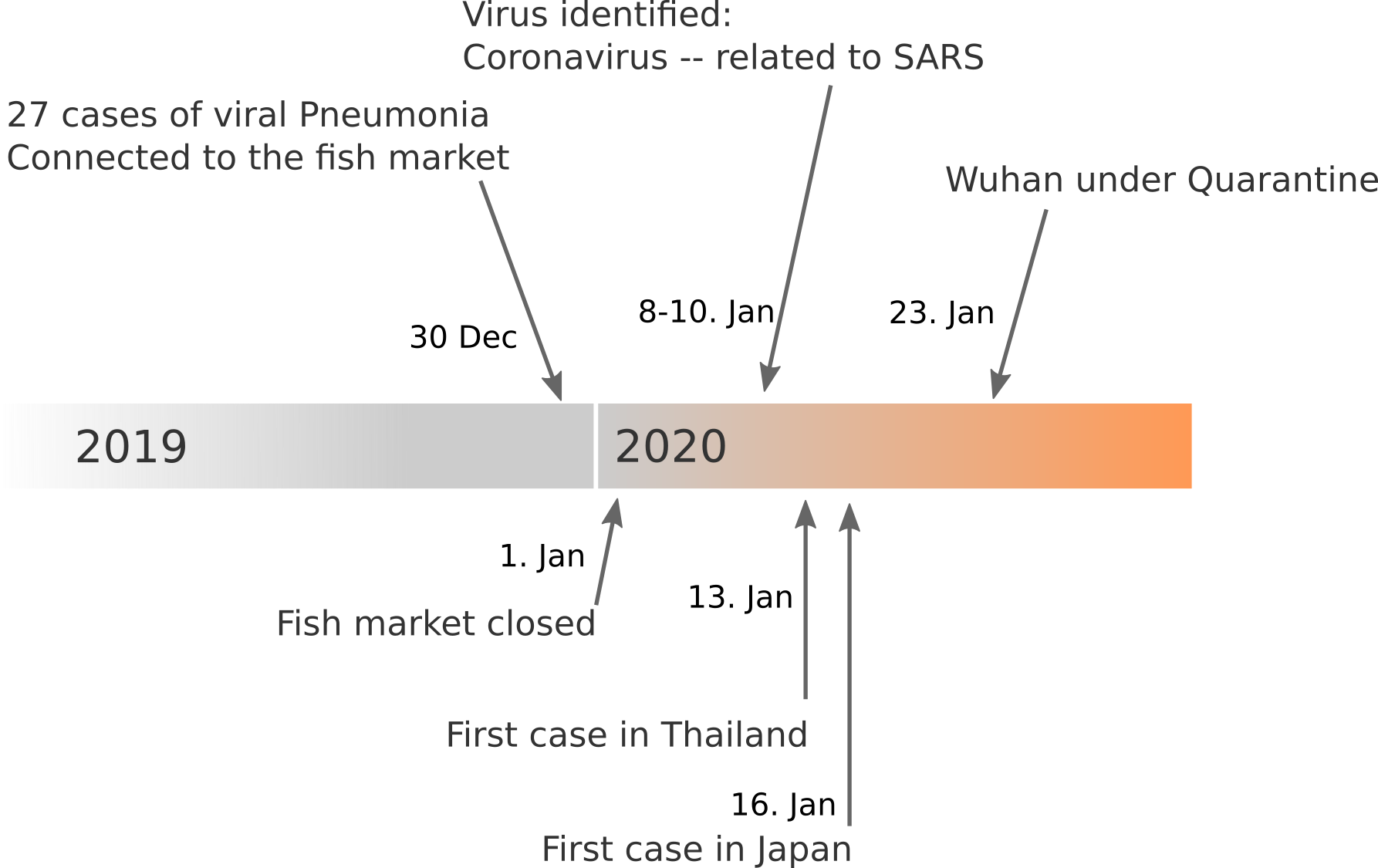 Data summarized by Ian MacKay
Data summarized by Ian MacKay
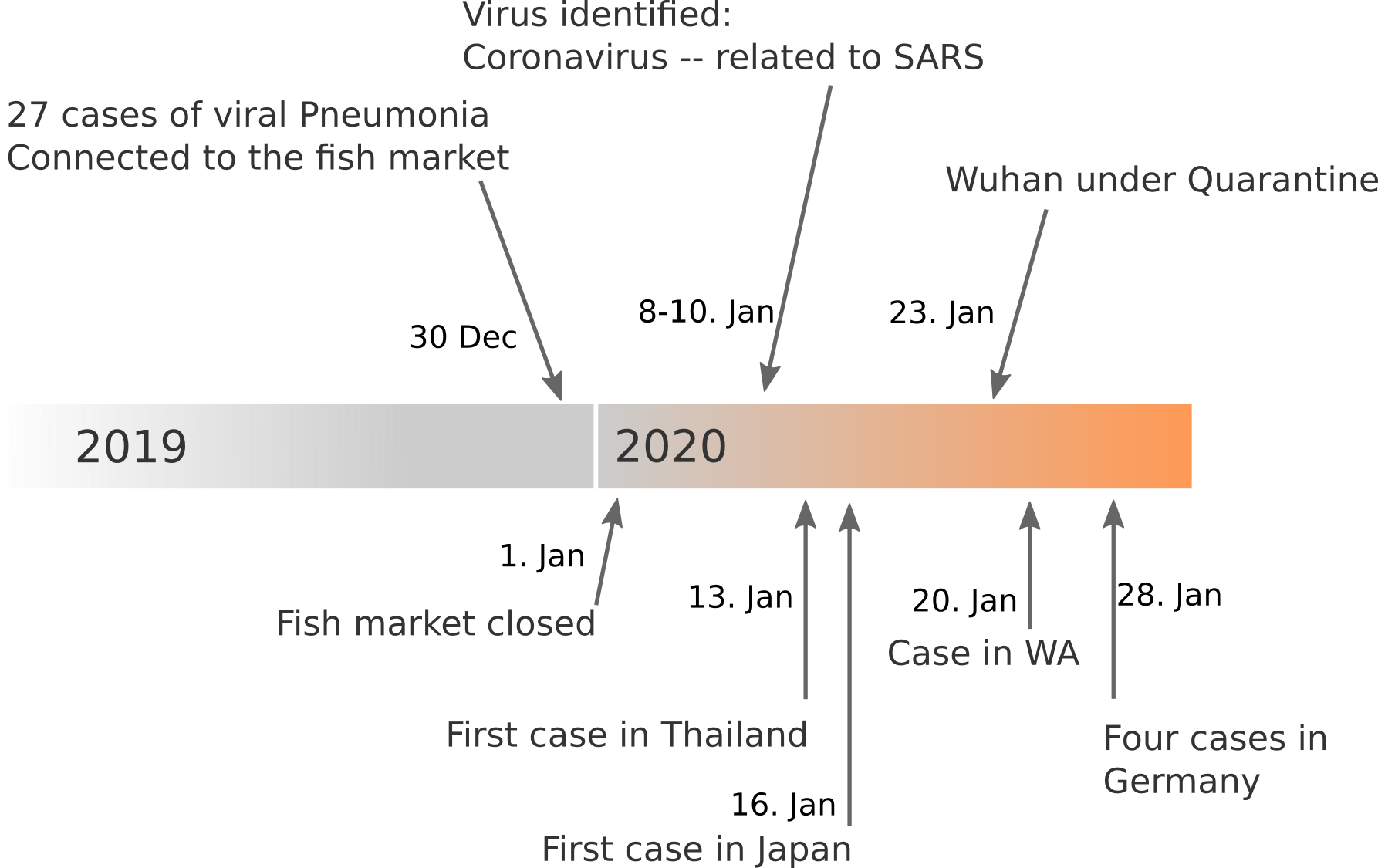 Data summarized by Ian MacKay
Data summarized by Ian MacKay
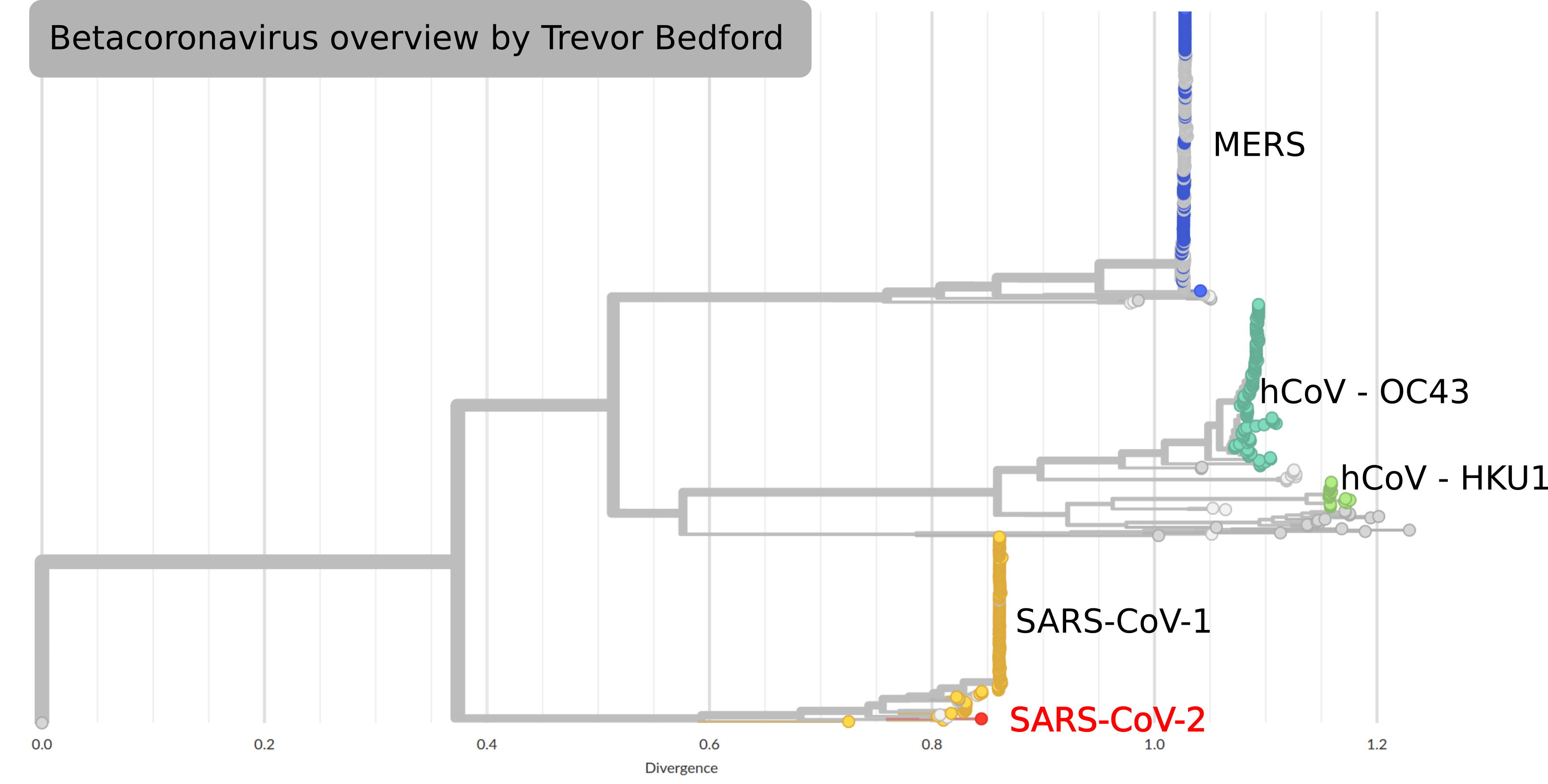 by Trevor Bedford
by Trevor Bedford
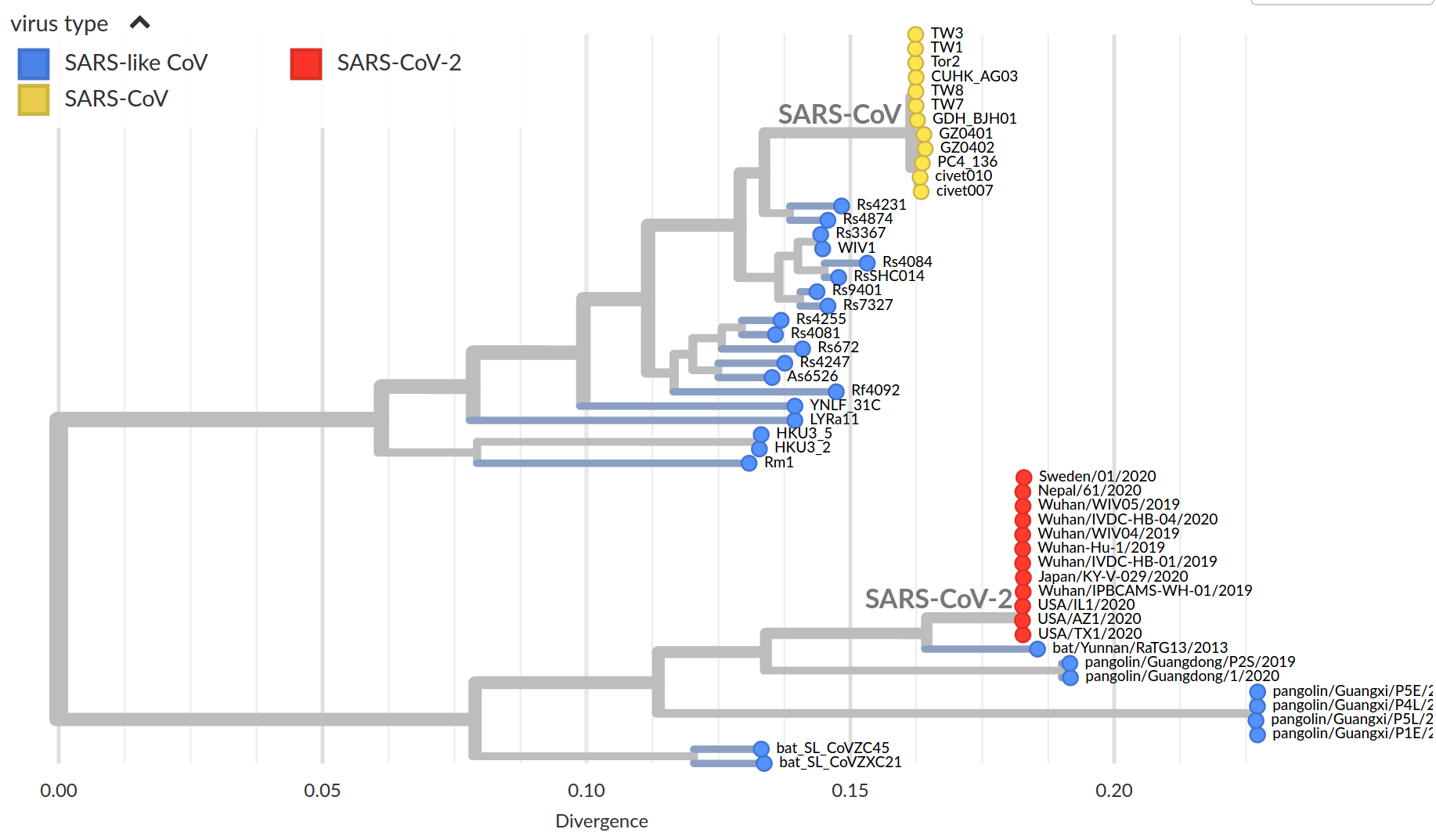 by Trevor Bedford
by Trevor Bedford
The SARS-CoV-2 genome
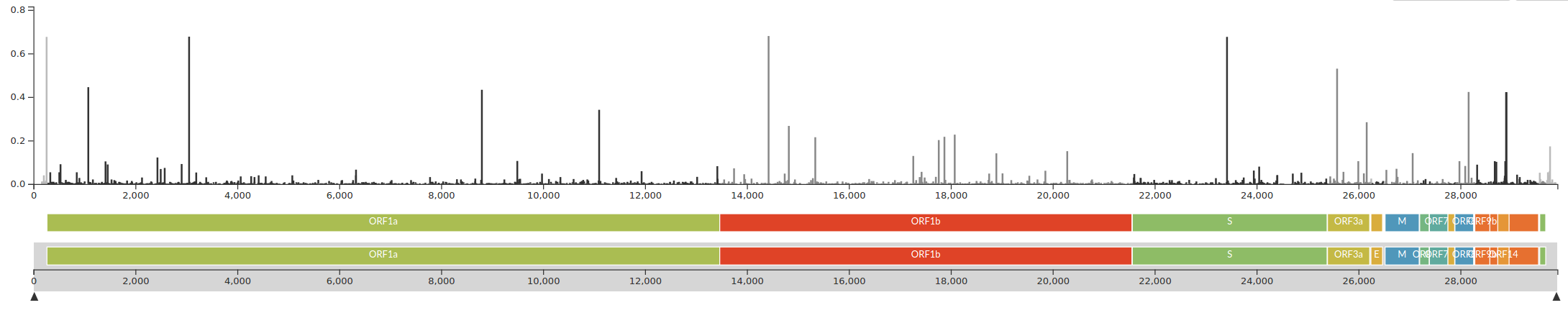
- 29k linear (+)ssRNA genome -- one of largest RNA virus genomes
- the first 2/3 code for the replication machinery
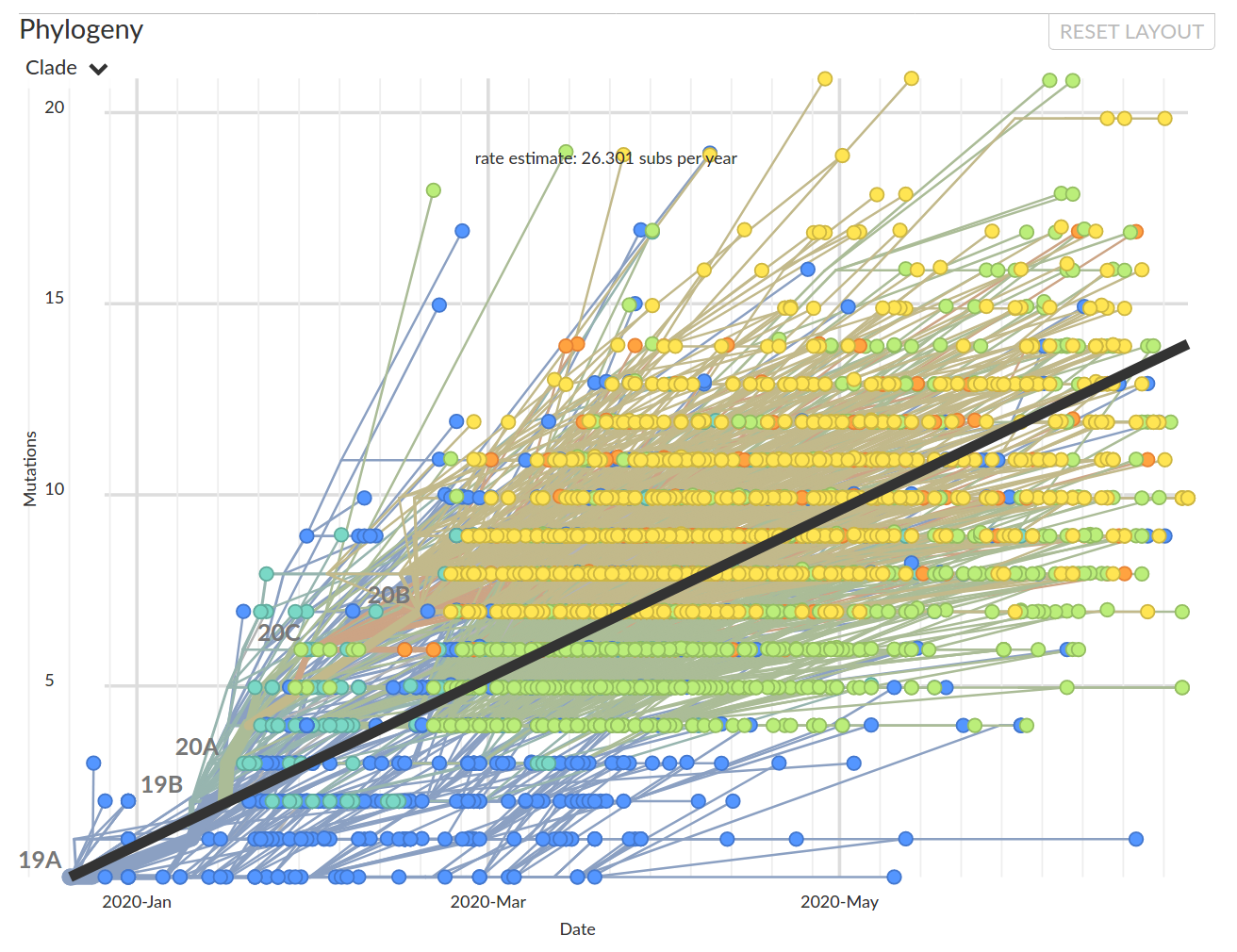
- While it spreads, it accumulates about 2 mutations per month
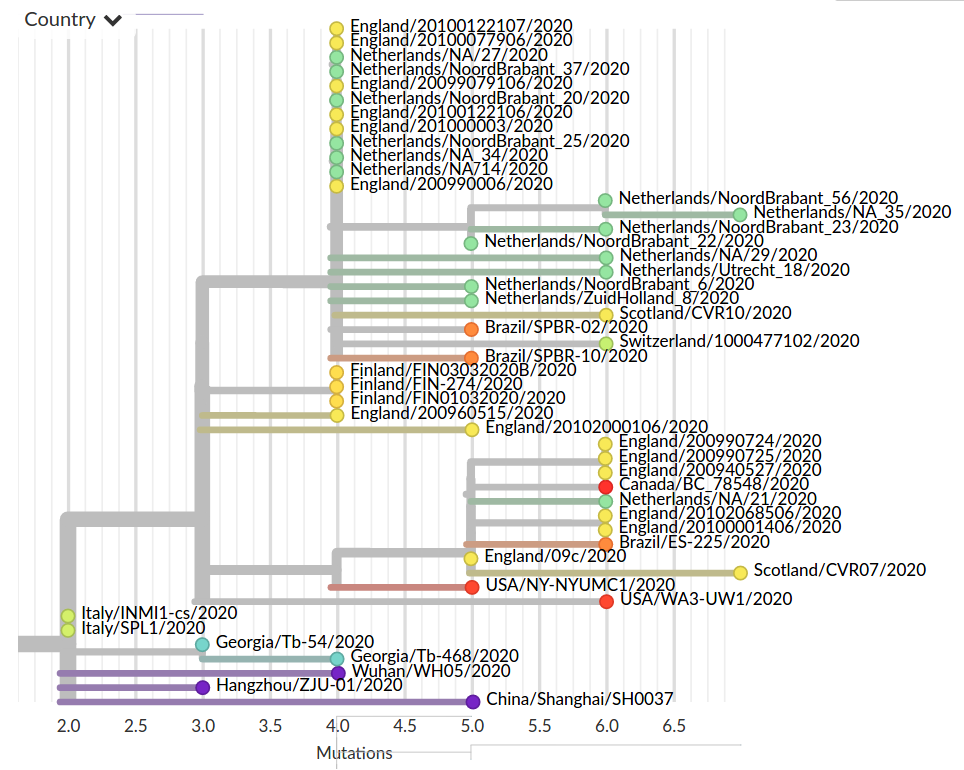
Tracking diversity and spread of SARS-CoV-2 in nextstrain
Available data on Jan 26
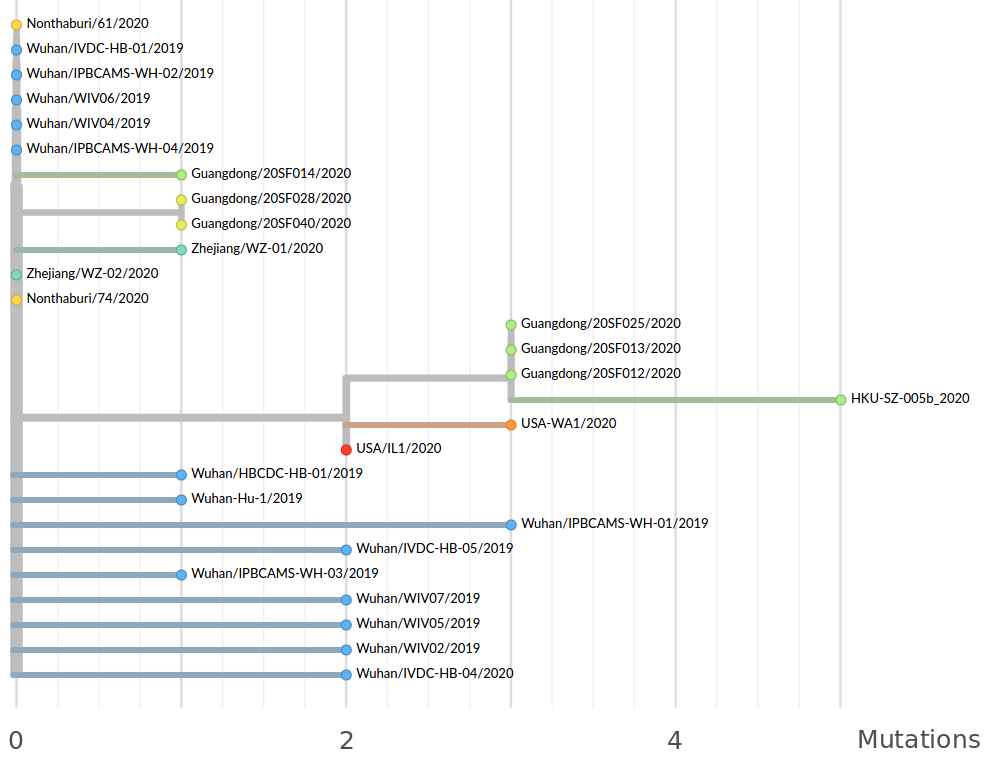
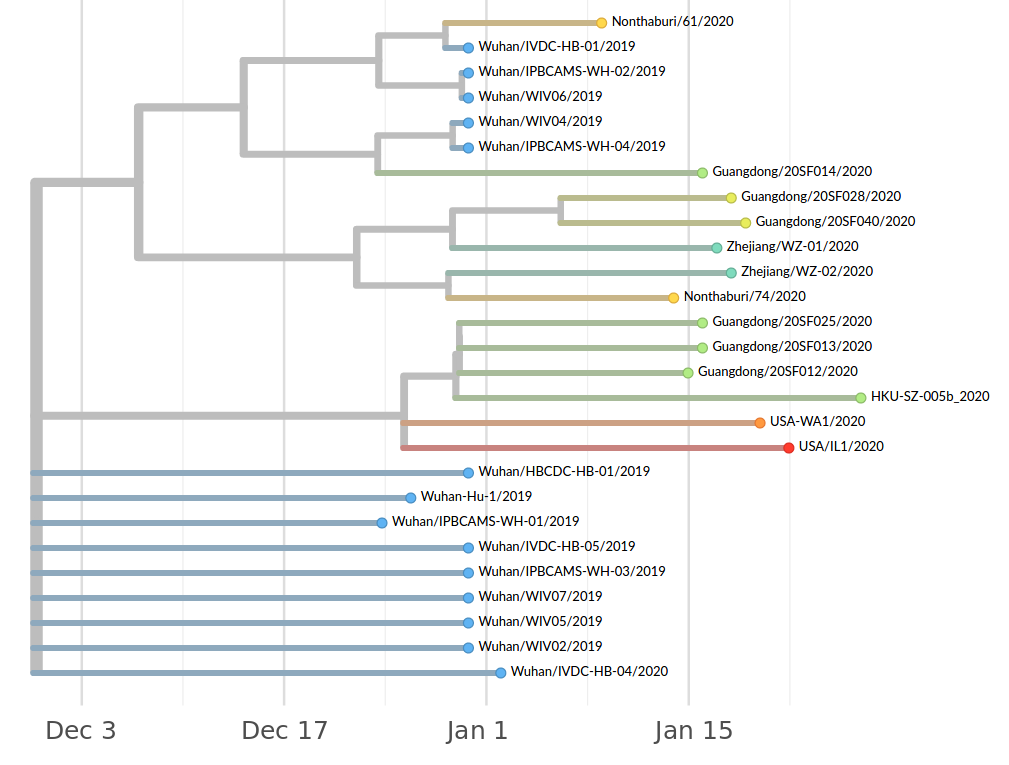
Early genomes differed by only a few mutations, suggesting very recent emergence
→ the closest to "real-time" we have experienced so far...
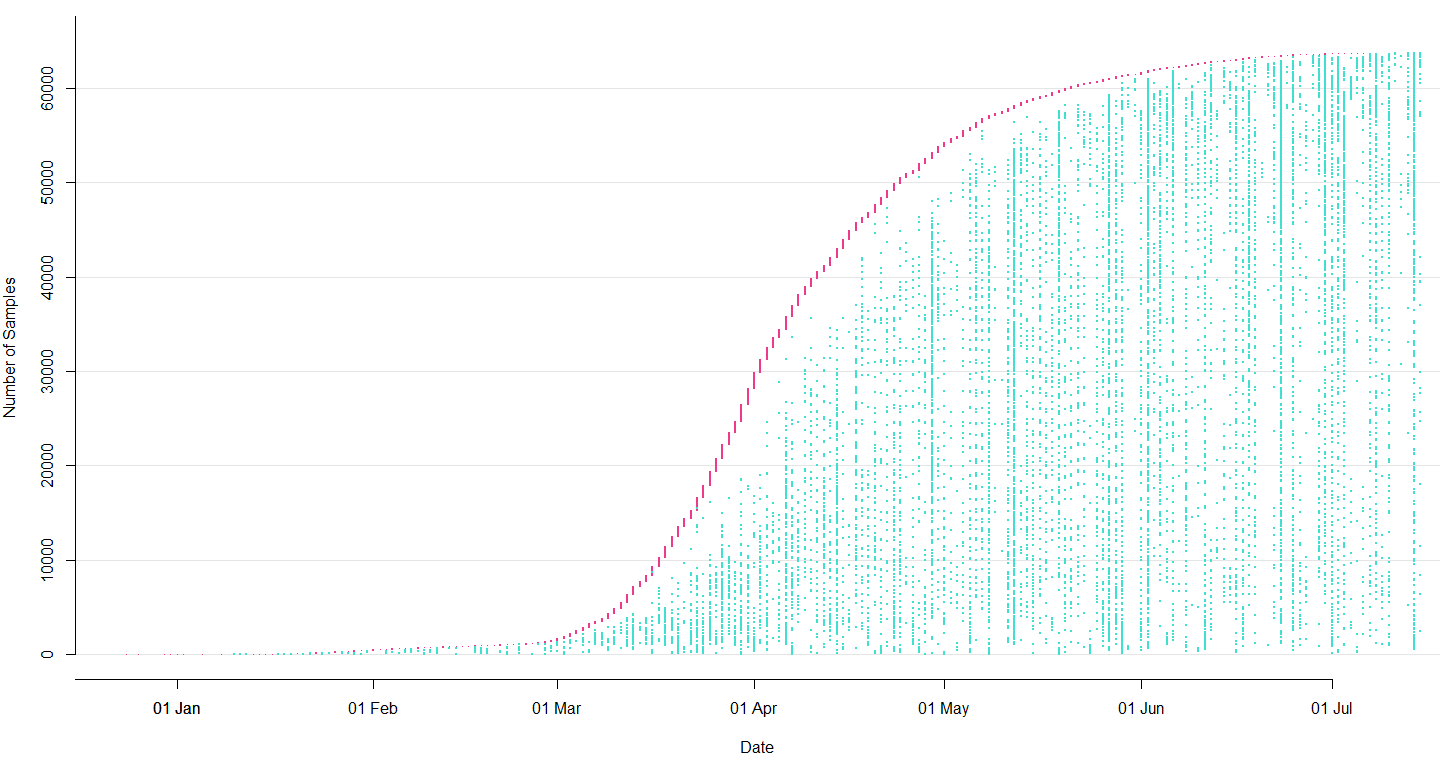
Figure by James Hadfield/Emma HodcroftSubset on July 14
As SARS-CoV-2 spreads, in accumulates on average 2 mutations per month
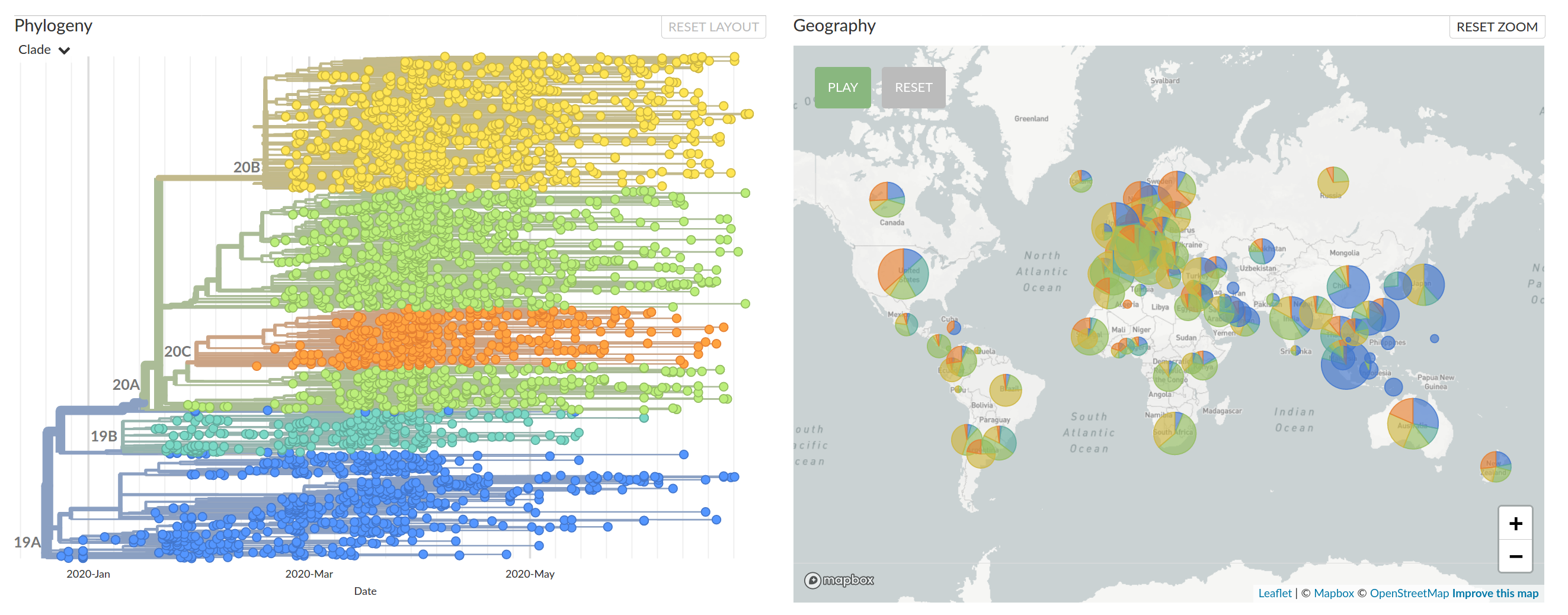
Tracking the spread of variants
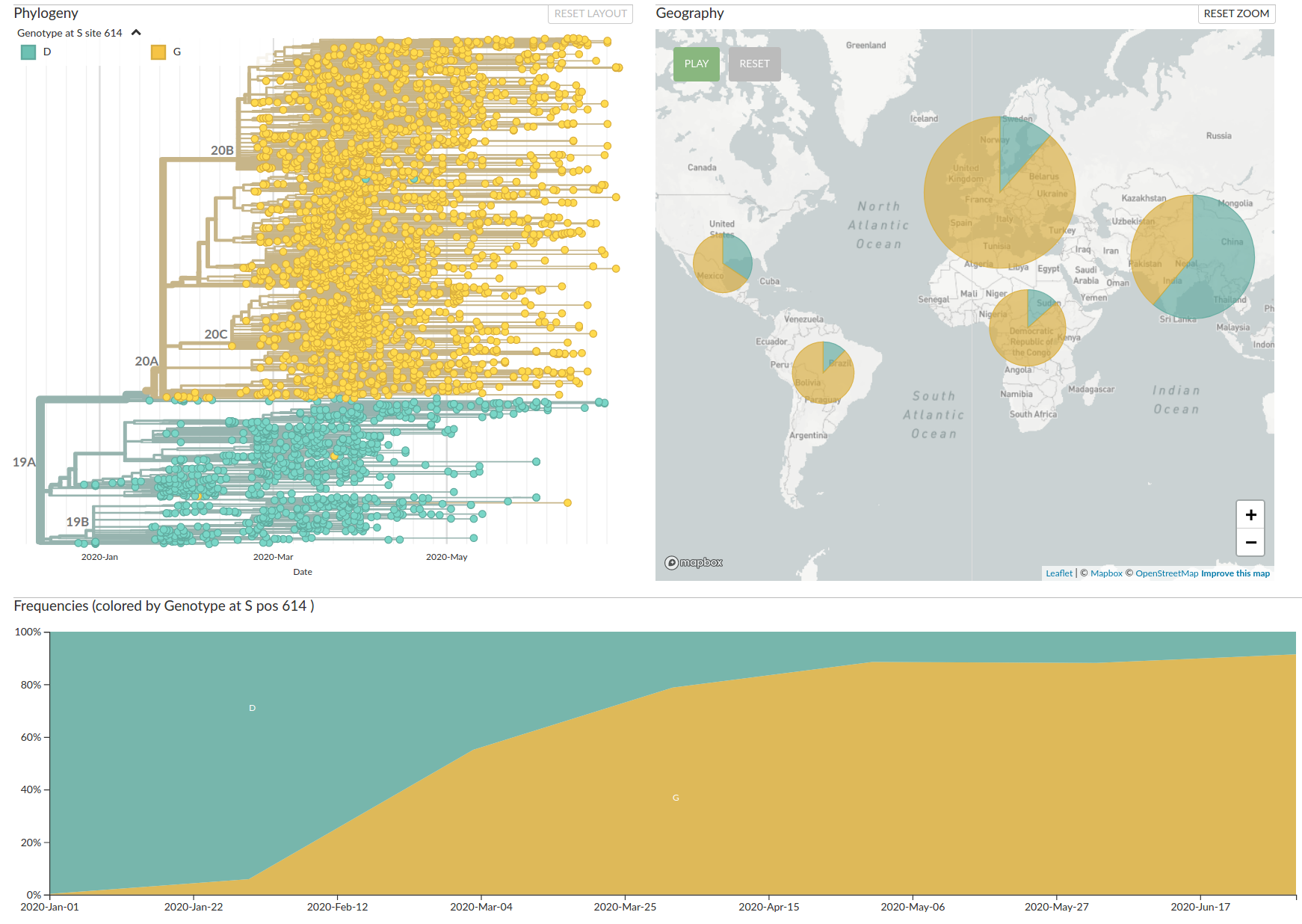
Tracking the spread of variants
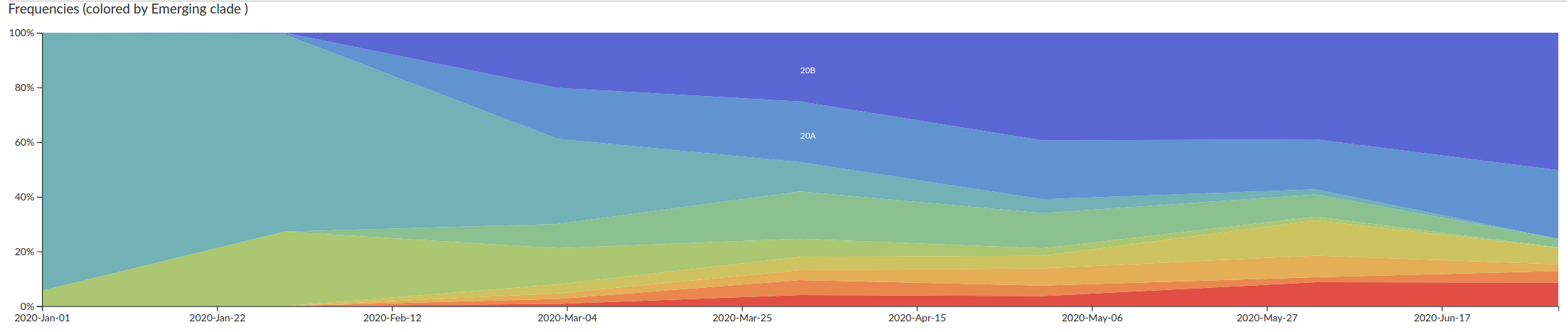
- Clade 20A (and its daughters 20B and 20C) have essentially taken over
- With them, several mutations, including S:D614G have become prevalent.
- There are reports that S:D614G increases transmission
(equivocal in my view) - Overall no strong signal that the virus has changed in meaningful ways
- But mutations are very useful to reconstruct how the virus spreads
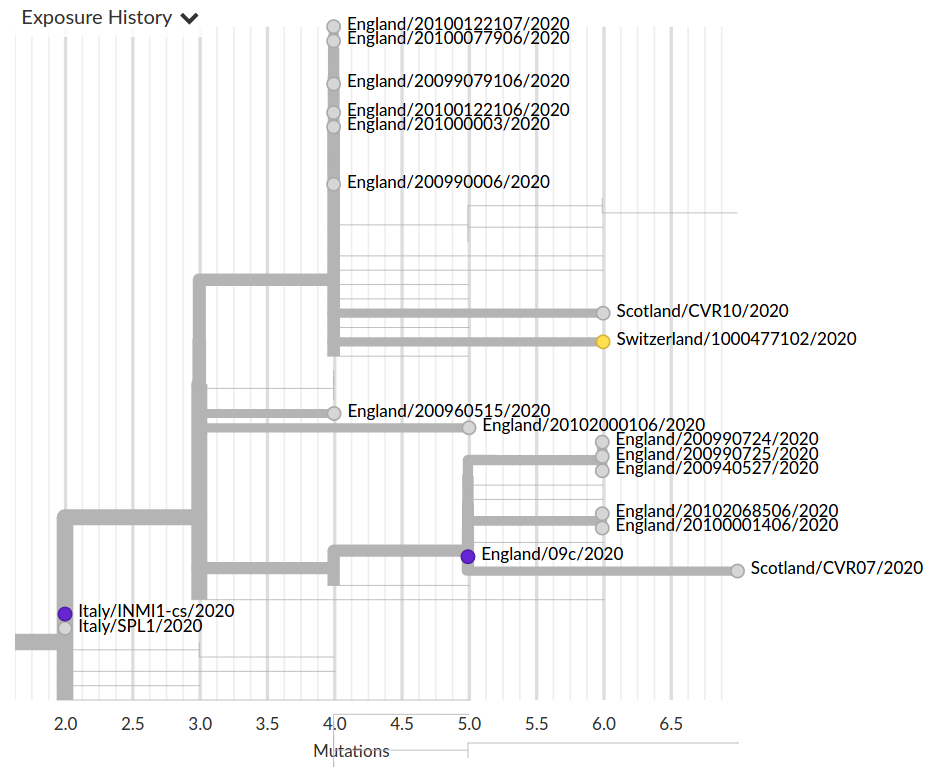
Genomic analysis as complement to contact tracing
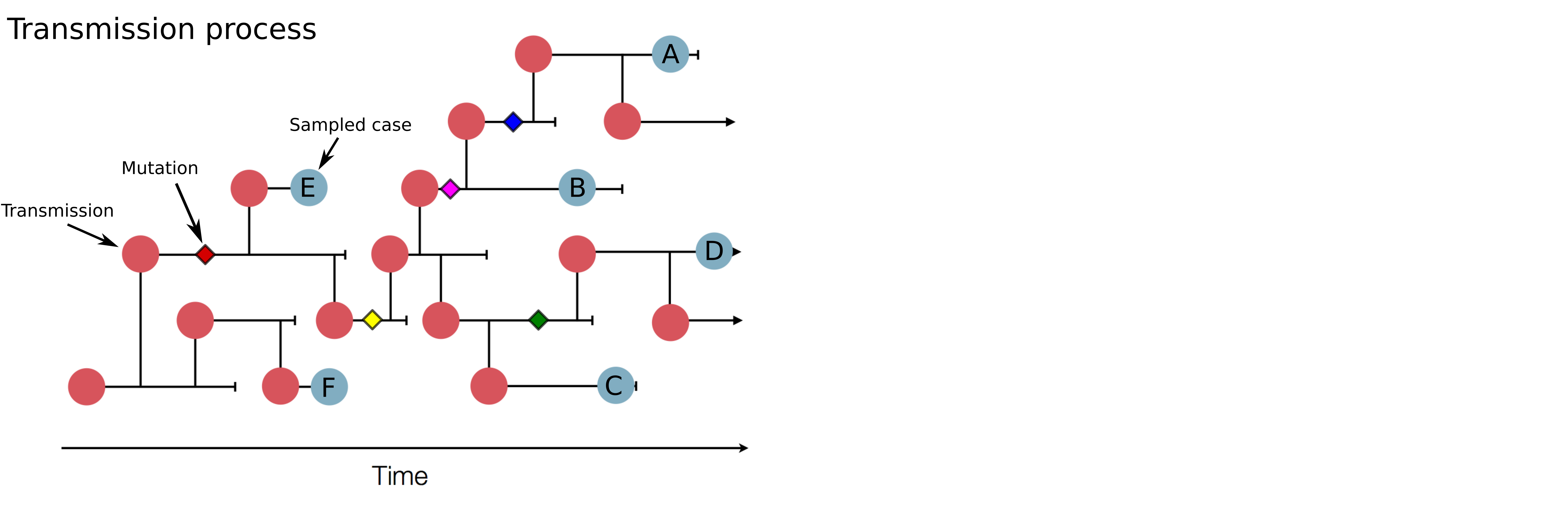
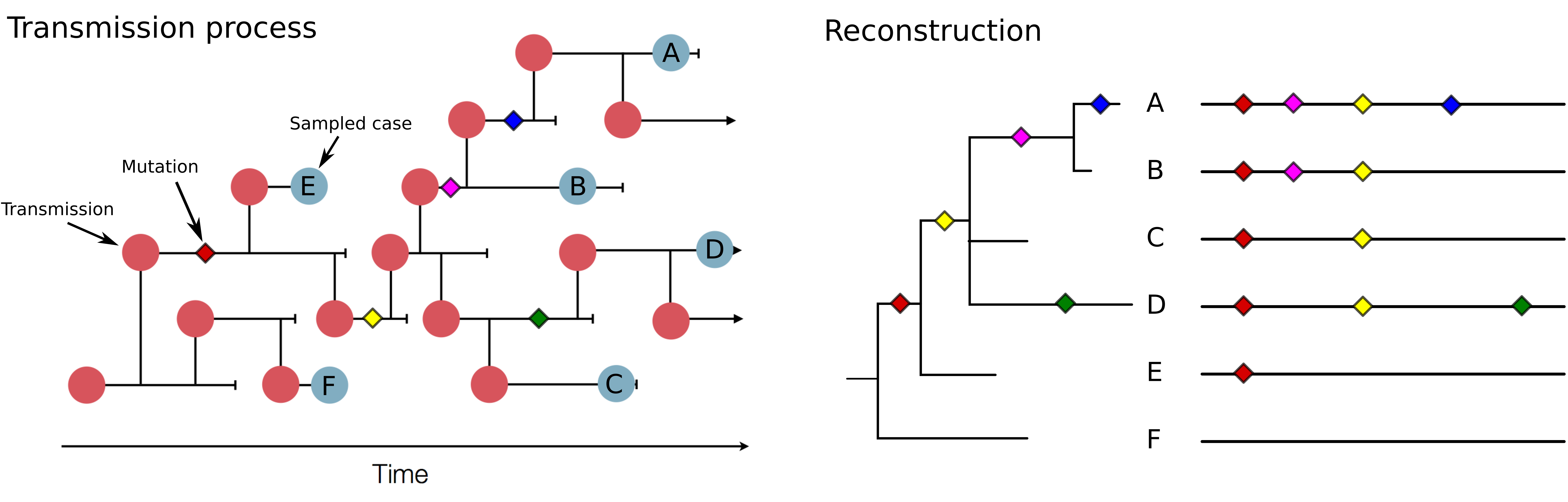
Tracing the origins of samples from Iceland
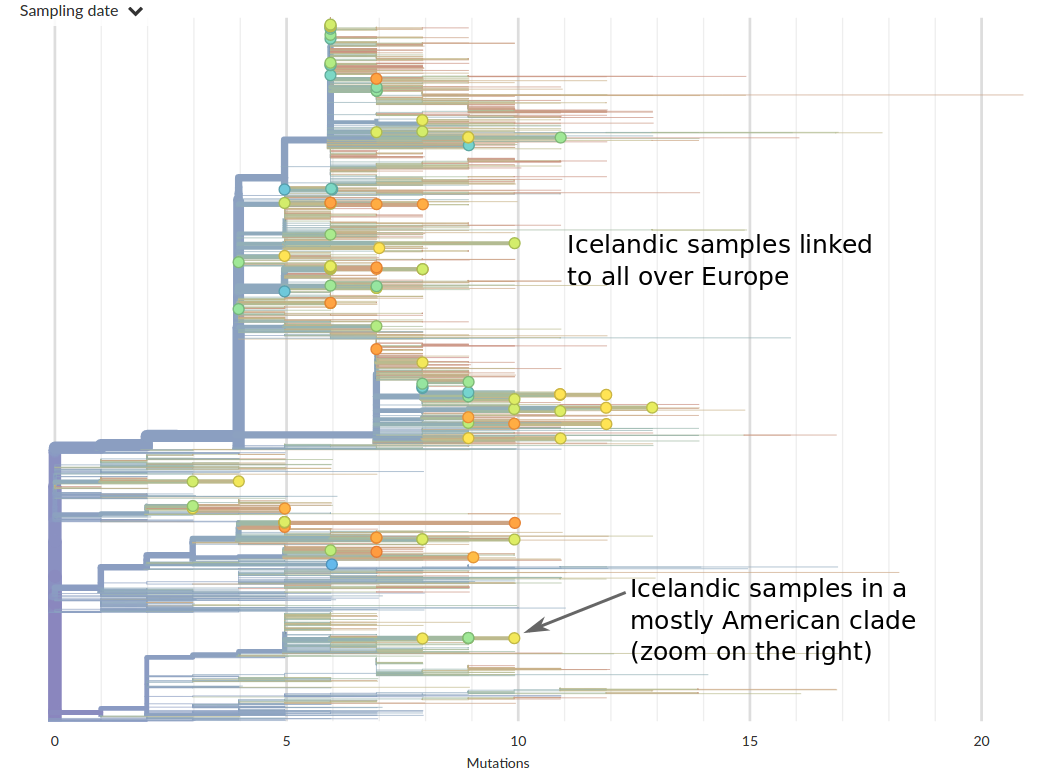
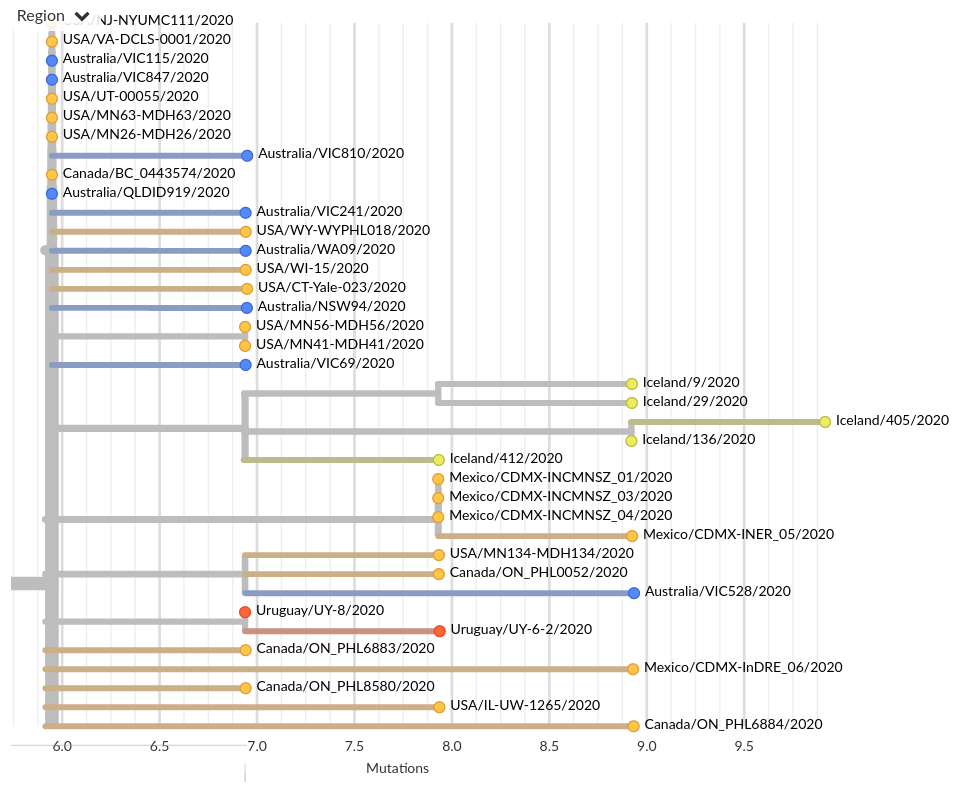
Prior to travel restrictions, SARS-CoV-2 is spread widely across the globe
SARS-CoV-2 phylodynamic analysis with nextstrain
- input: metadata (csv table) + sequences
- composable snakemake pipeline
- filtering (select relevant genomes)
- alignment
- tree building
- infer time scaled phylogenies
- ancestral state reconstruction and phylogeography
- clade classification
- export to visualization
- runs in 1-3h for 4k genomes
Visualization with nextstrain
- run locally (localhost)
- deployed on your own servers
- shared via github hosting
- shared via nextstrain group (aws S3)
- sensitive metadata can be dropped onto the tree
- browser based tools to analyze your own sequences
Seasonal incidence of influenza viruses
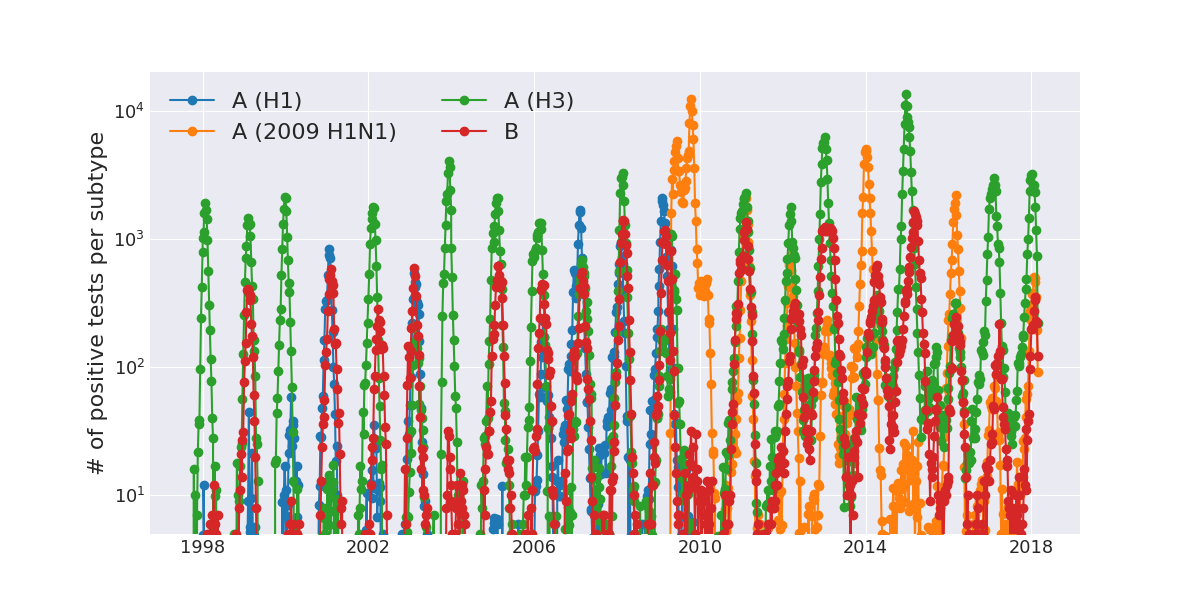
2009 pandemic influenza -- UK
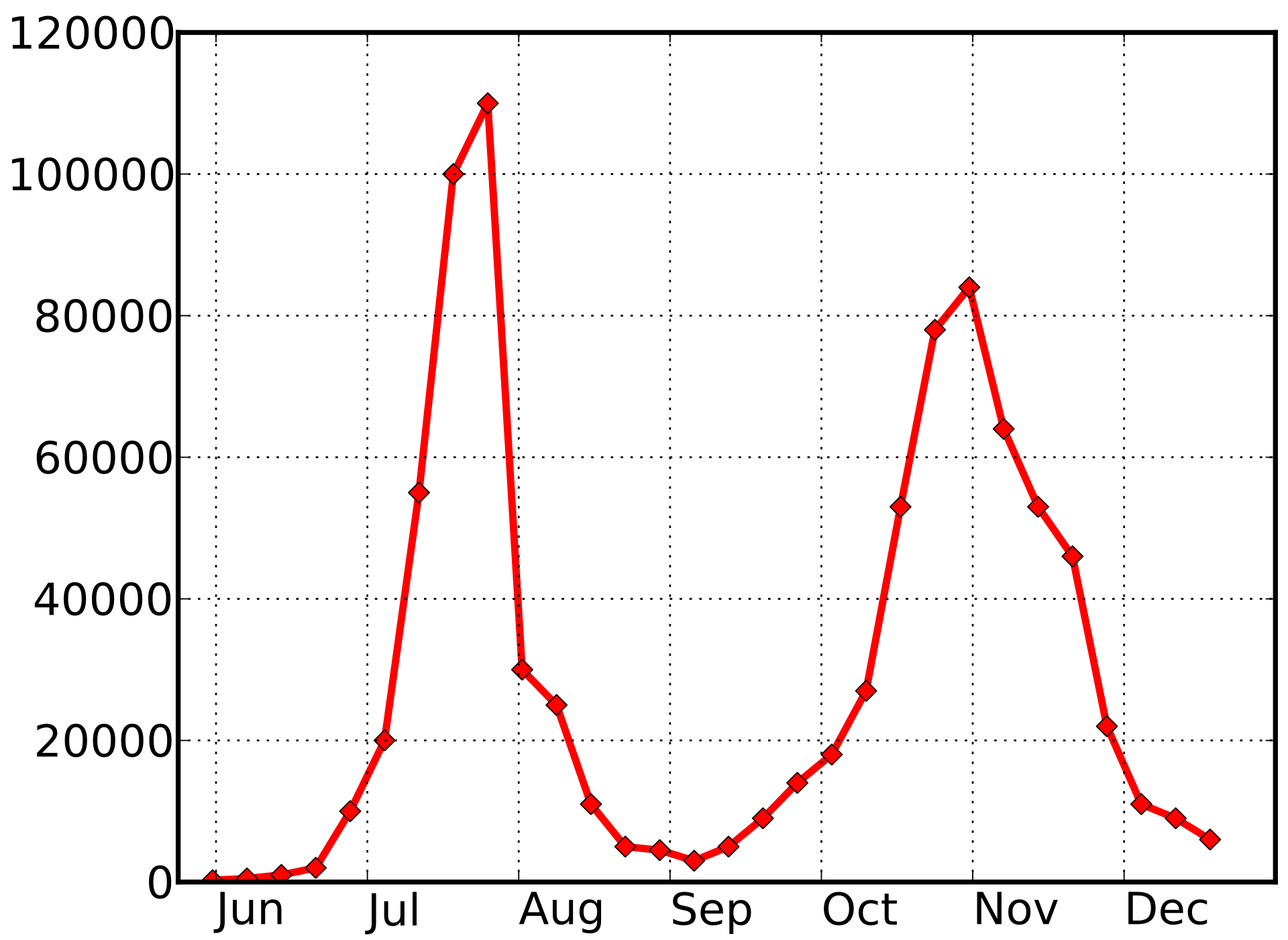
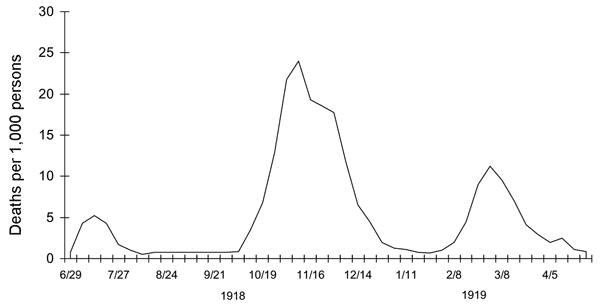
1918 influenza --- UK
 by Trevor Bedford
by Trevor Bedford
Human corona viruses have pronounced seasonal prevalence (Sweden)
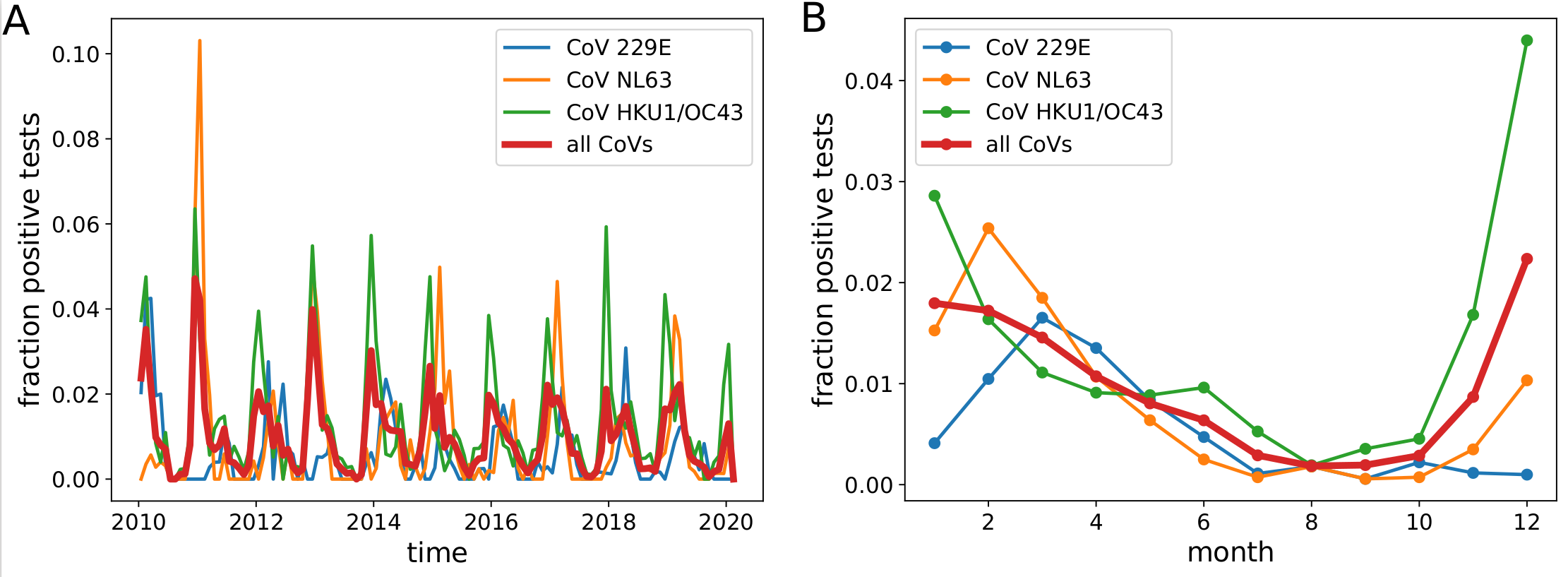
- Respiratory virus incl seasonal CoVs show seasonality
- Forcing through human behavior (indoor/outdoor activities).
- Control of the virus might be harder in temperate winter
- Absolute effect of seasonality unknown
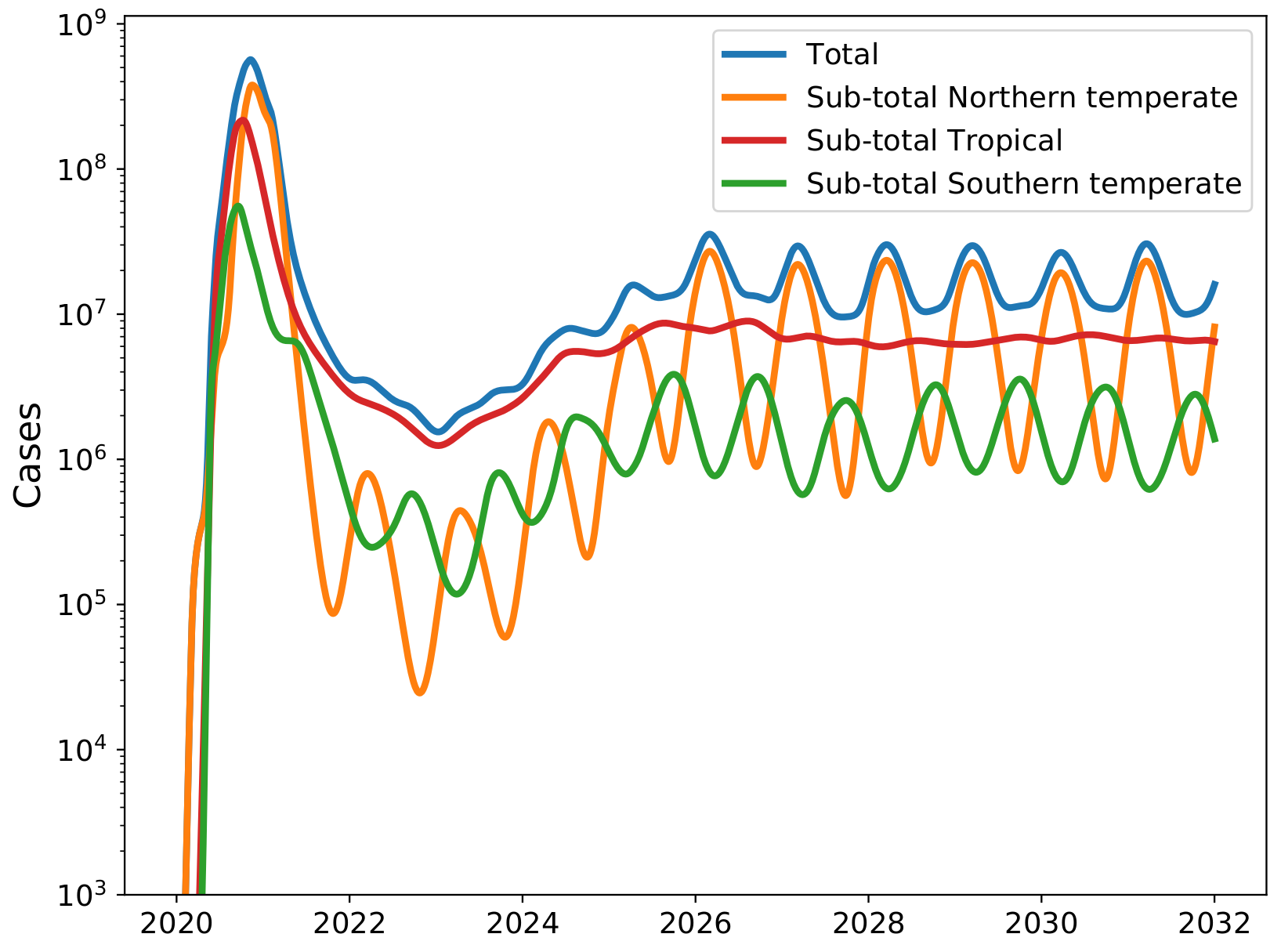
Potential transition to an endemic seasonal virus
Acknowledgments
Trevor Bedford and his lab -- terrific collaboration since 2014
especially James Hadfield, Emma Hodcroft, Ivan Aksamentov, Moira Zuber, and Tom Sibley
Data we analyze are contributed by scientists from all over the world
Data are shared and curated by GISAID
Acknowledgments
- Robert Dyrdak
- Jan Albert
- Valentin Druelle
- Emma Hodcroft
Fighting misinformation
Colorful trees are easily misinterpreted...