Reconstructing, tracking, and predicting viral spread and evolution
Richard Neher
Biozentrum & SIB, University of Basel
slides at neherlab.org/202204_Ascona.html
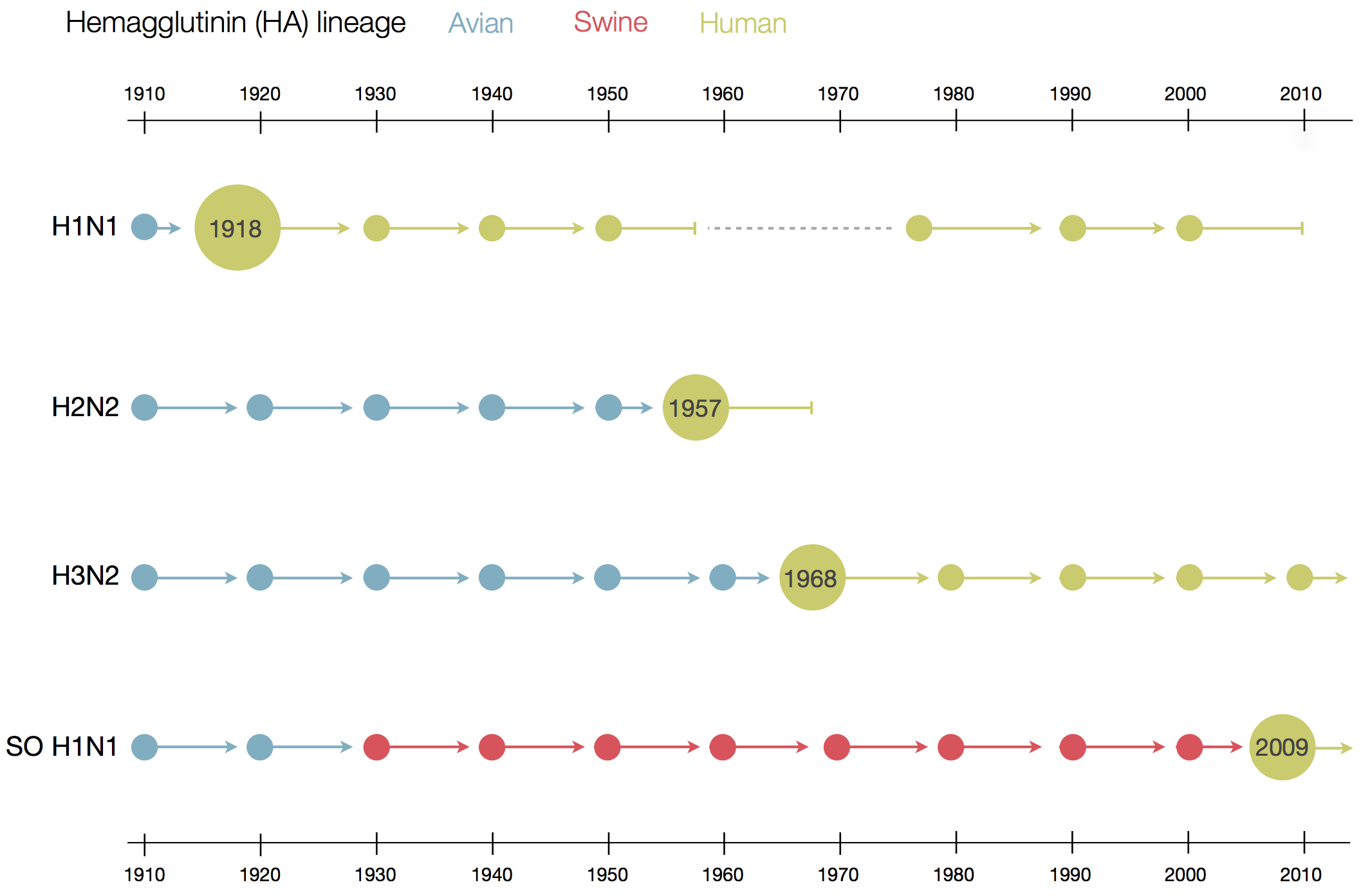
Human seasonal influenza viruses
Positive tests for influenza in the USA by week
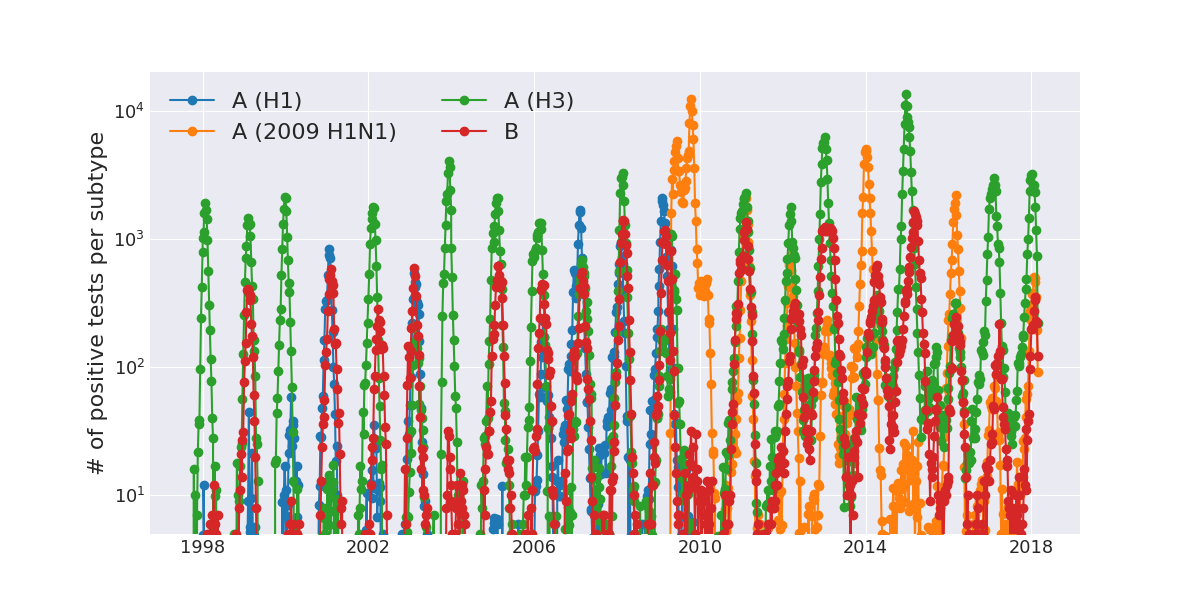 Data by the US CDC
Data by the US CDC
Human corona viruses are seasonal (Sweden)
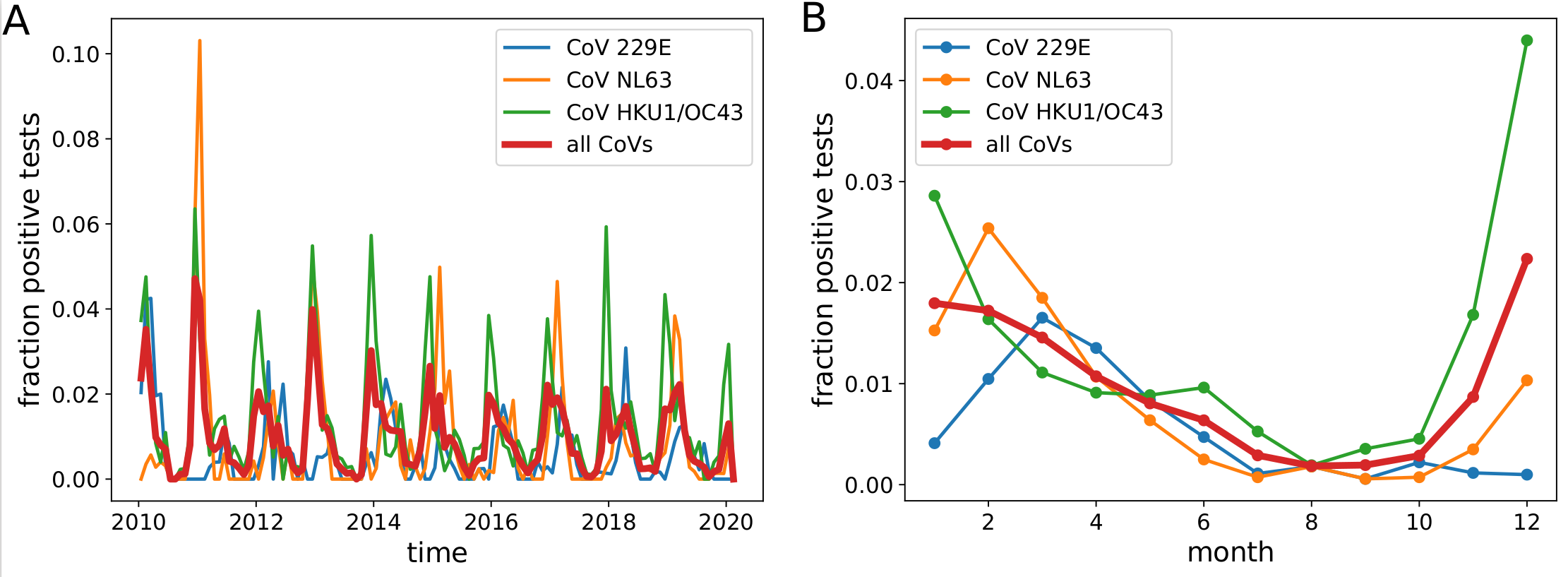
- Respiratory virus incl seasonal CoVs show seasonality
- Forcing through human behavior (indoor/outdoor activities).
- Absolute effect of seasonality unknown
- The two alpha and beta coronaviruses alternate
Viral dynamics beyond case counts?
Genomic analysis to reconstruct pathogen spread and evolution
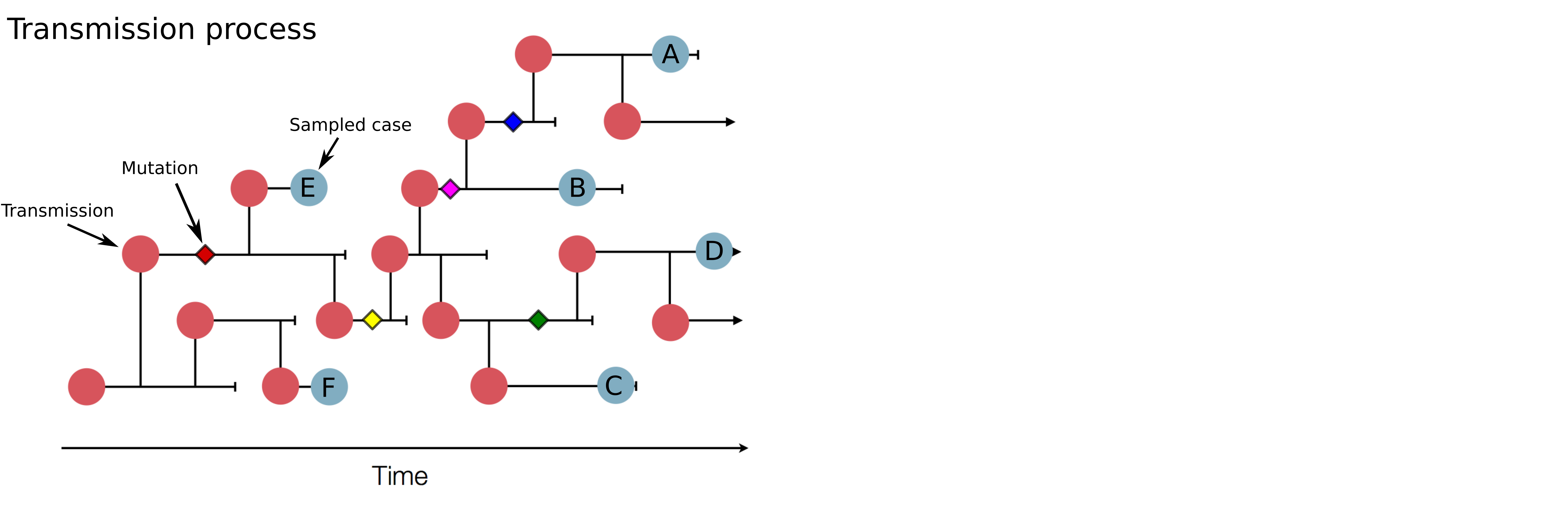
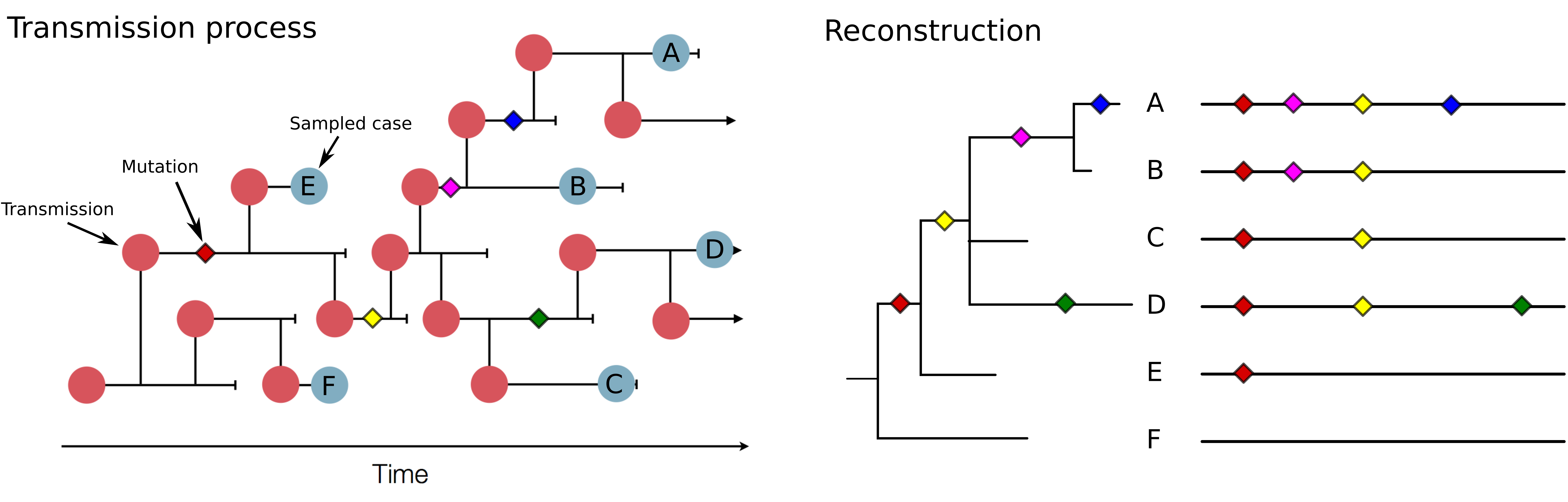
Mutations allow to reconstruct how pathogens spread -- clusters, introductions, etc
Mutations can result in phenotypic change -- immune escape, drug resistance, host adaptation
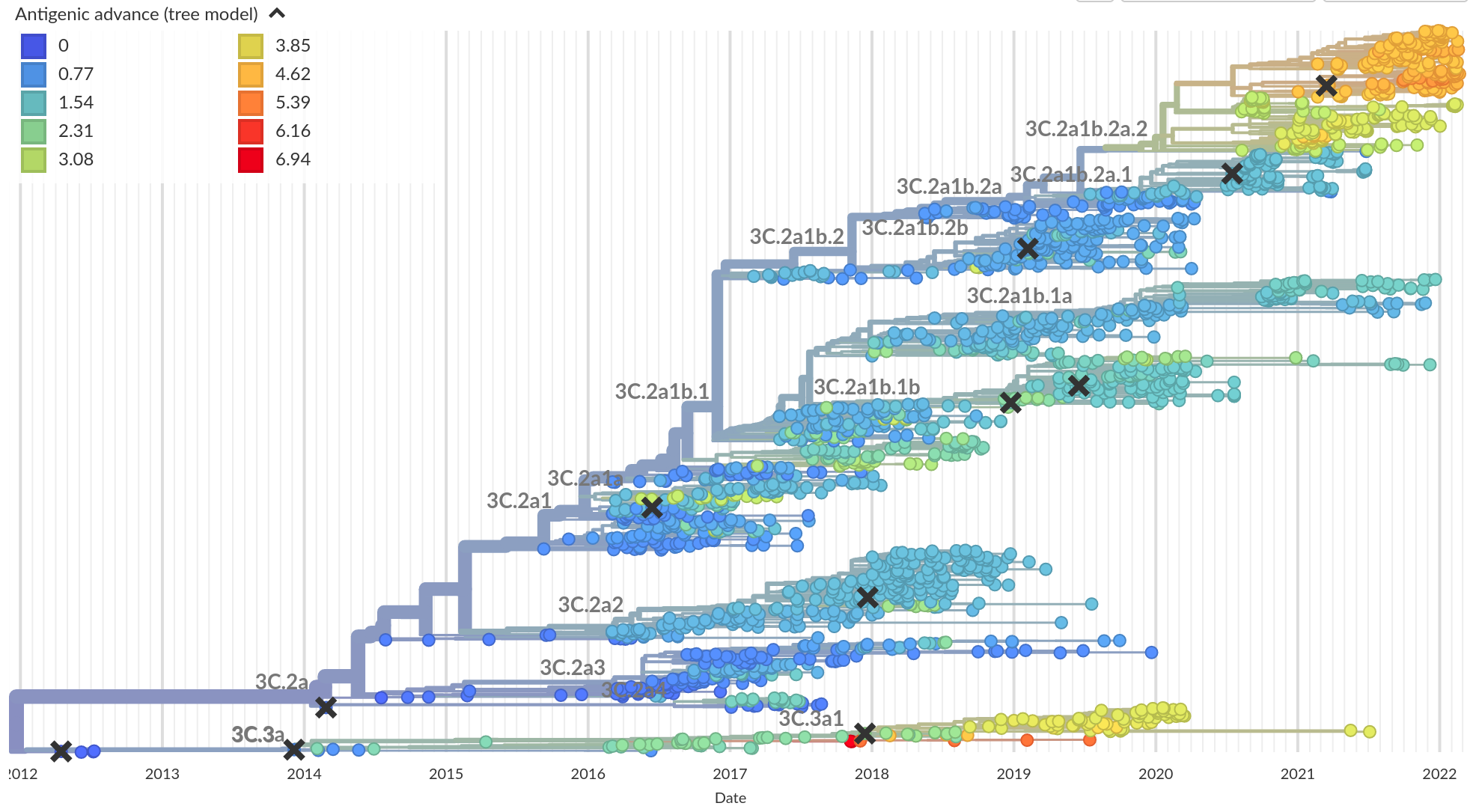
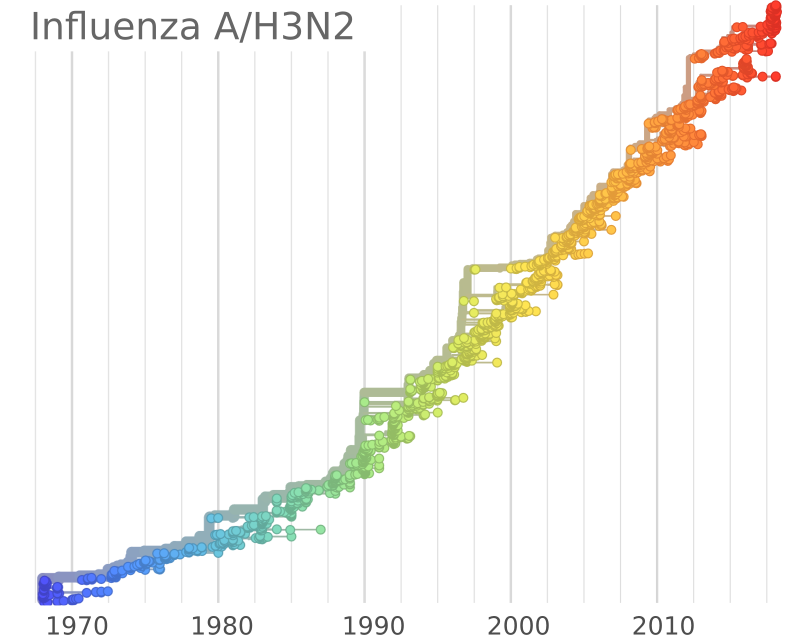
Influenza A/H3N2
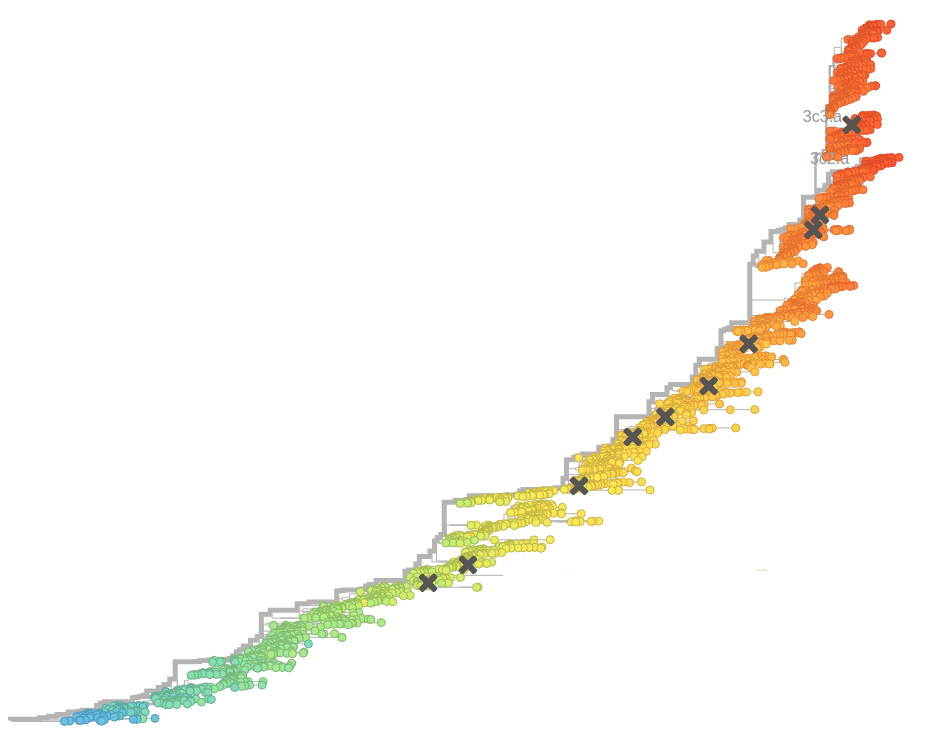
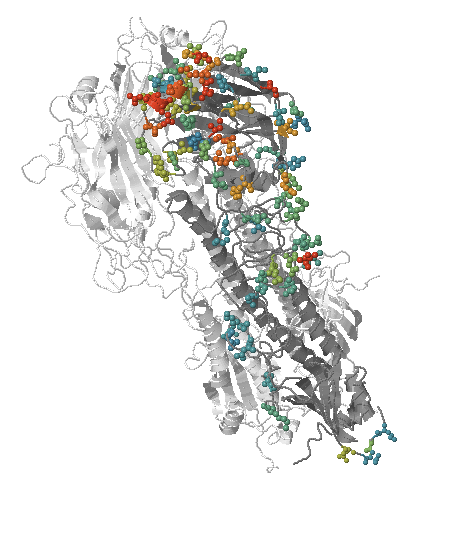
- Influenza viruses evolve to avoid human immunity
- Vaccines need frequent updates
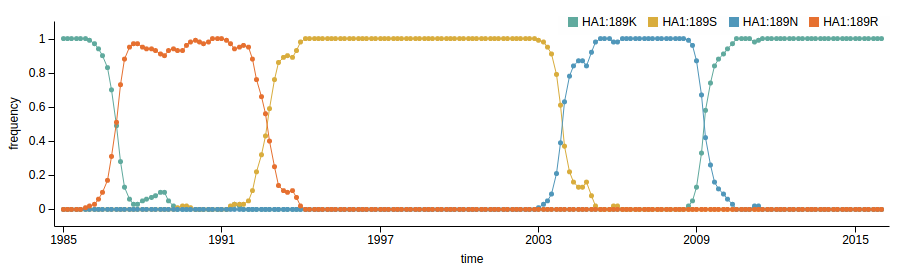
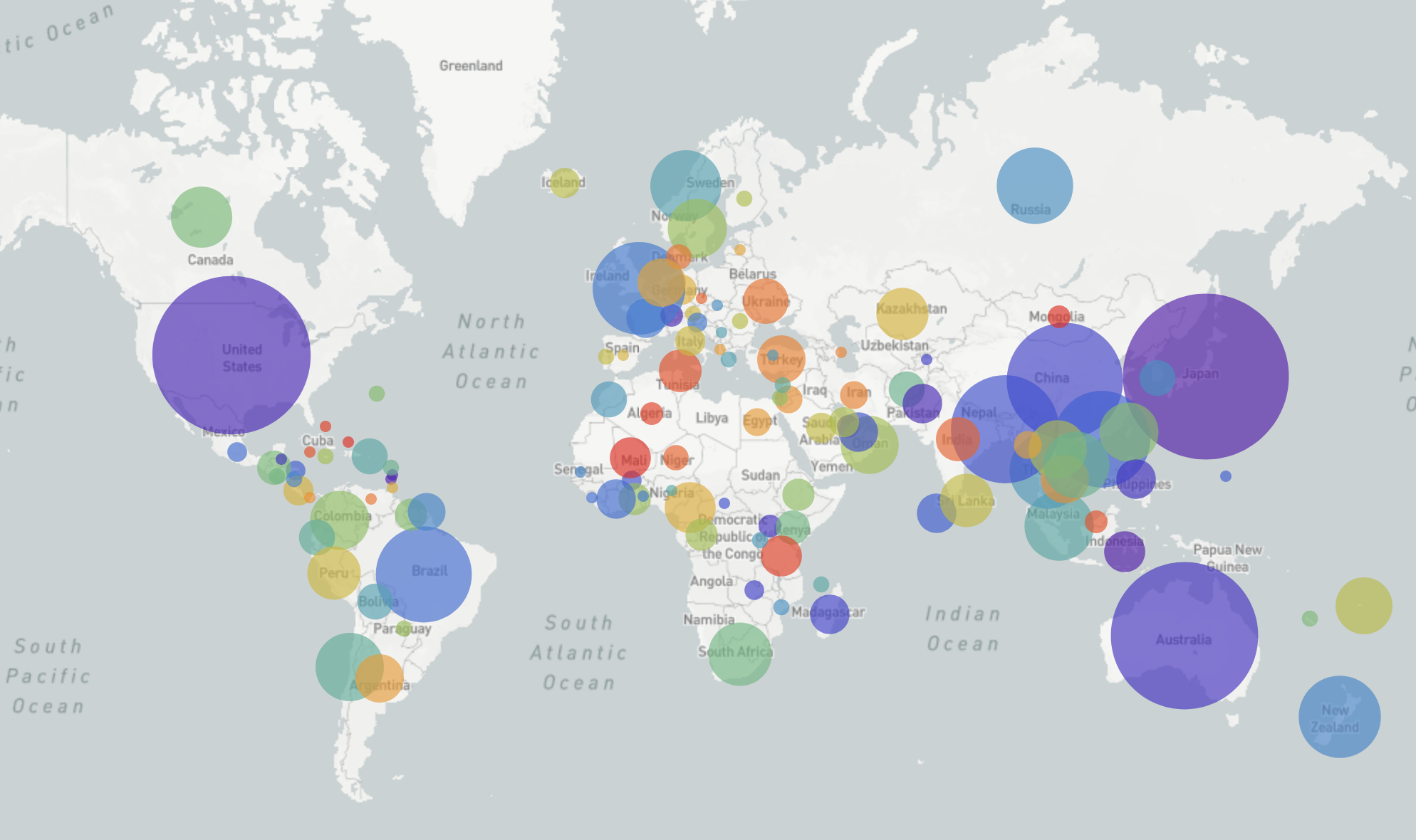
nextflu.org
joint project with Trevor Bedford & his lab
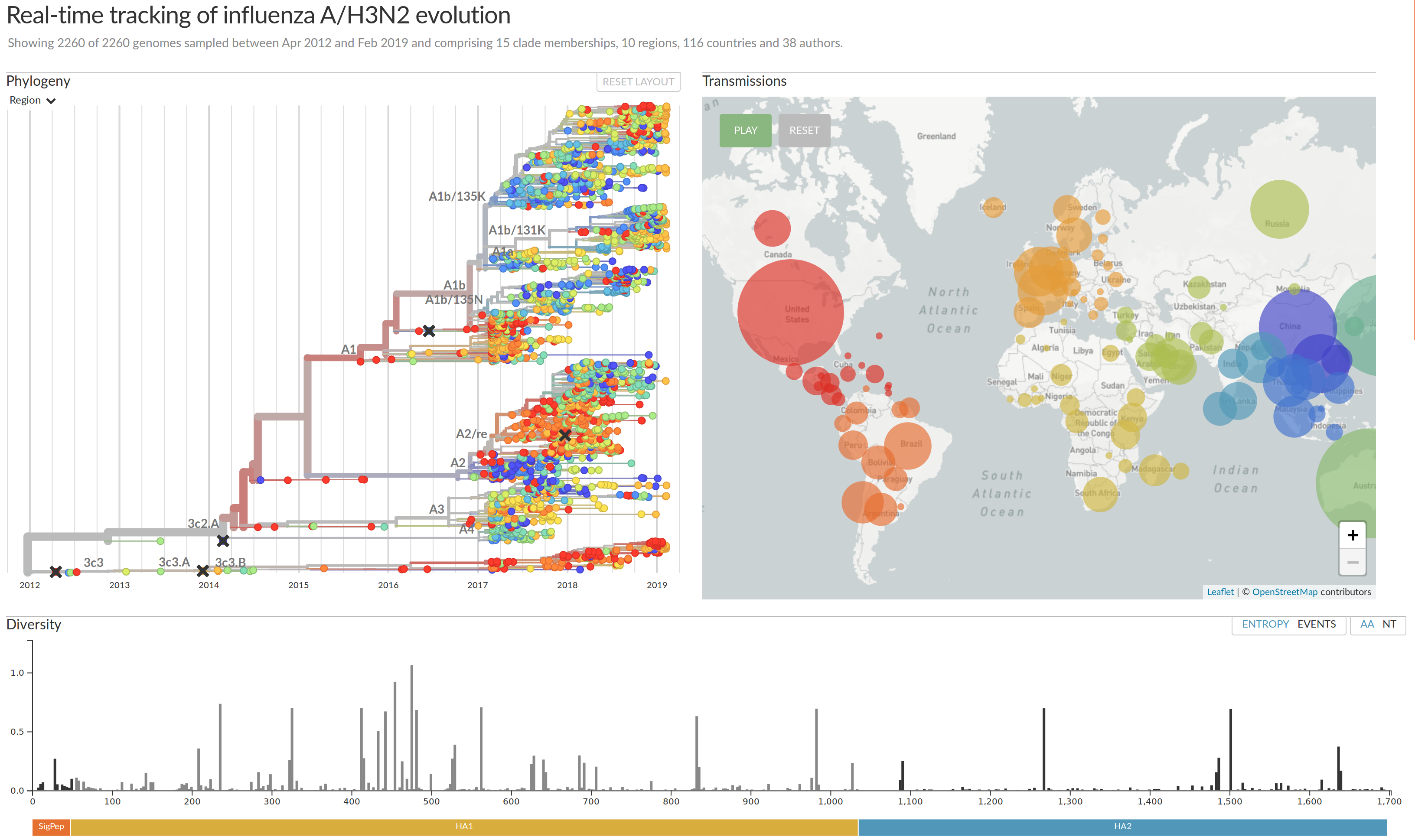
Beyond tracking: can we predict?
Prediction of the dominating H3N2 influenza strain
- Explicit fitness scores based on specific mutations
→ mutations in previously characterized epitopes
→ mutations that likely reduce fitness
→ mostly historically ascertained - Phylogenetic indicators to spot rapidly expanding clades
- Laboratory data (antigenicity, virulence)
- Human serology
- (other models try to predict incidence, not the dominating strain)
Mutational signatures -- epitope mutations
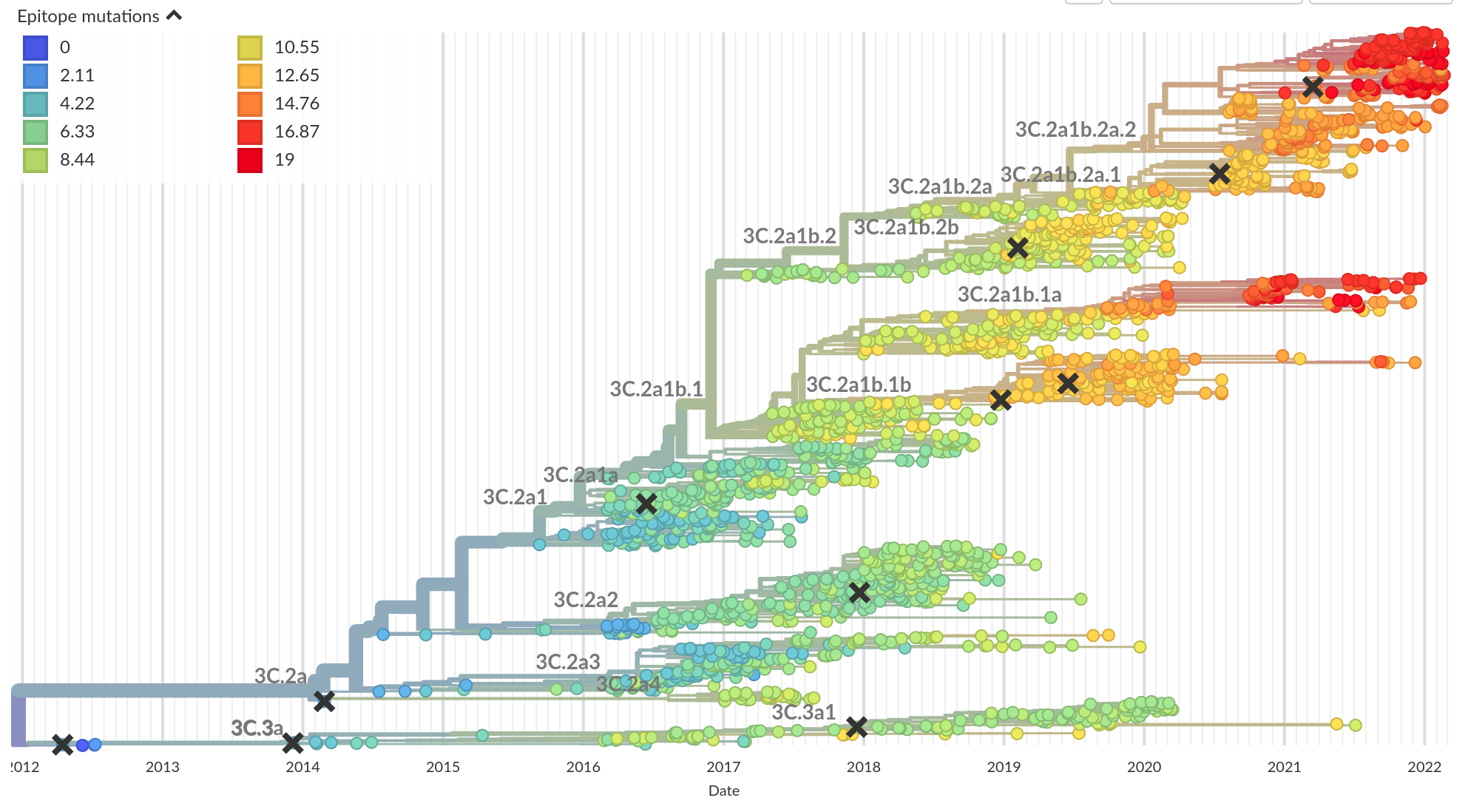 Luksza and Laessig, Nature, 2014
Luksza and Laessig, Nature, 2014
Phylogenetic indicators -- LBI
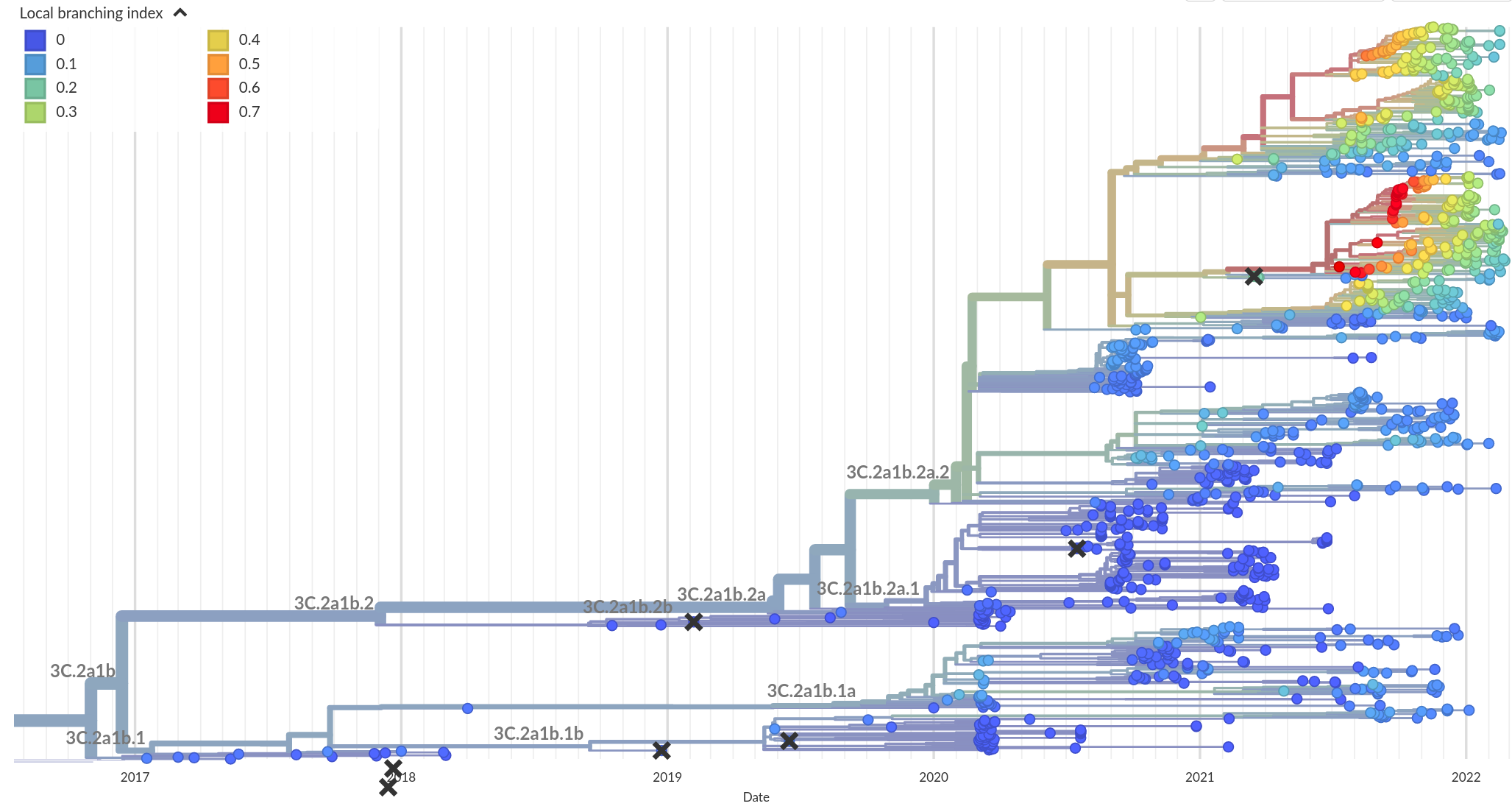 RN, Russell, Shraiman, eLife, 2014
RN, Russell, Shraiman, eLife, 2014
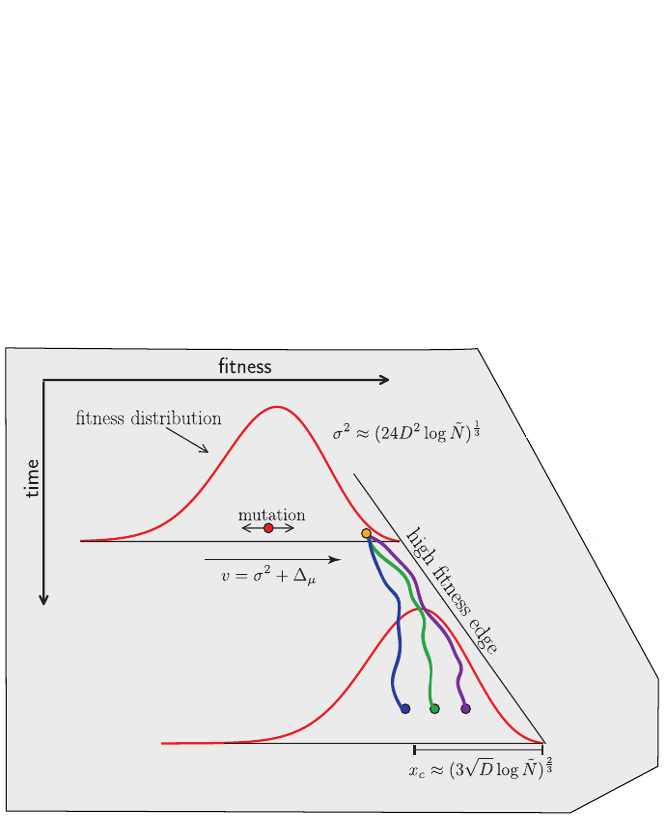
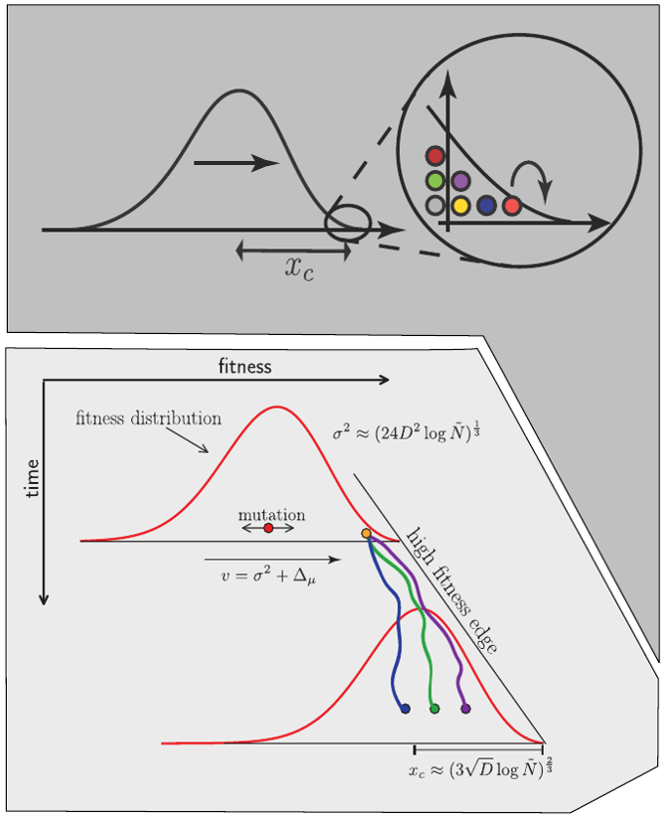
Fitness variation in rapidly adapting populations
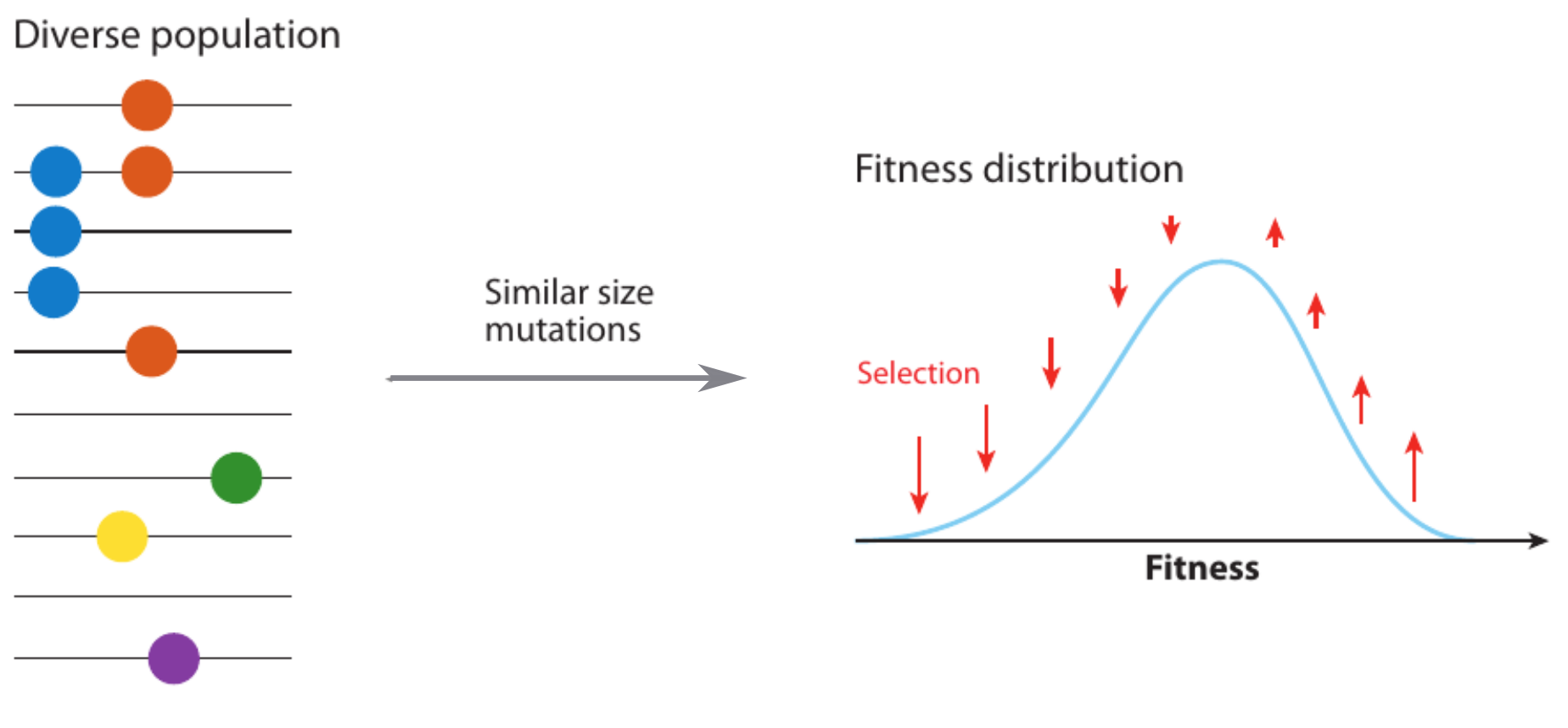
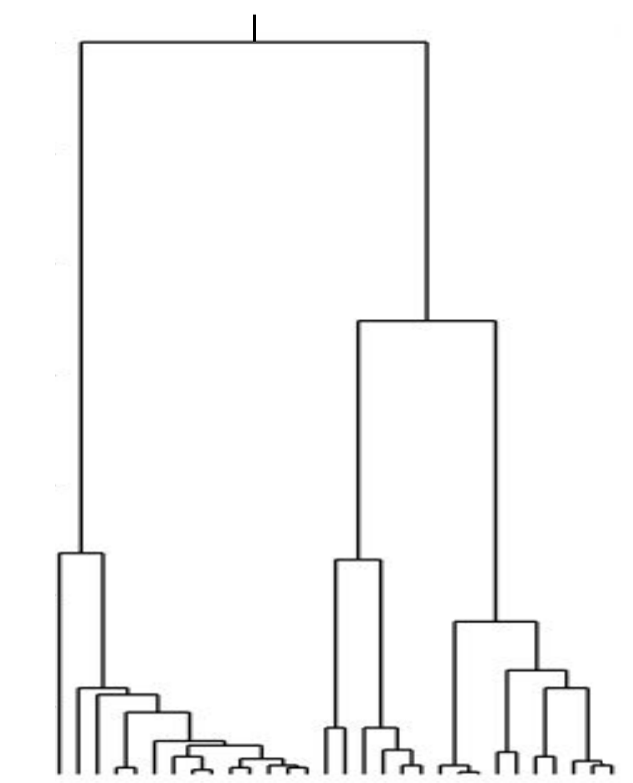
Neutral/Kingman coalescent
strong selection
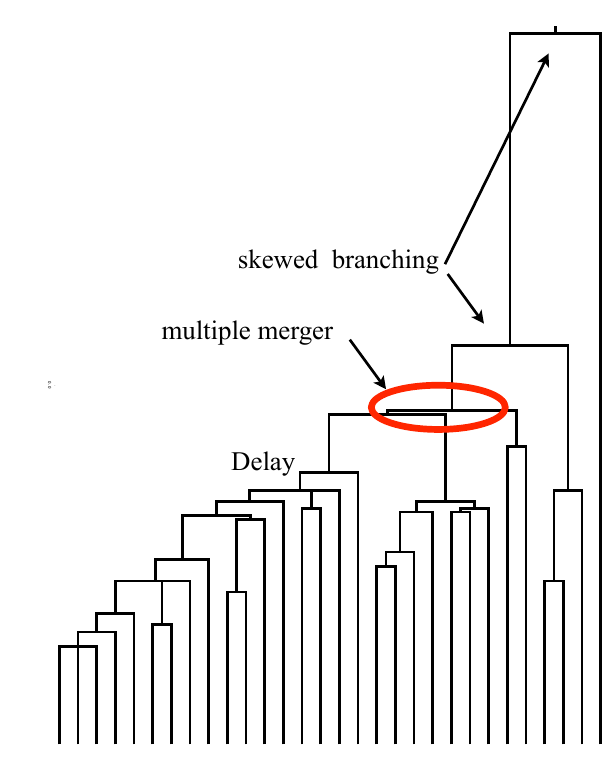
Bolthausen-Sznitman Coalescent
Hemagglutination Inhibition assays
Antigenic distance tables
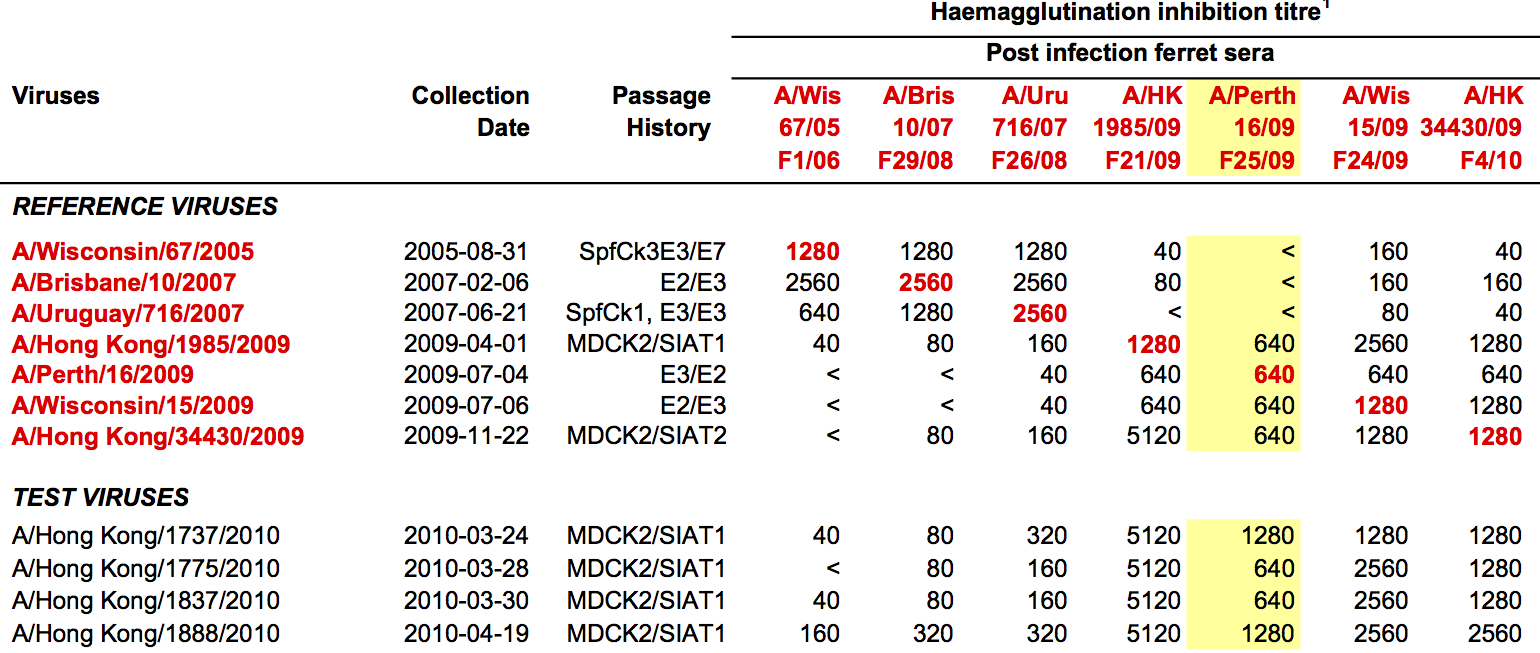
- Long list of distances between sera and viruses
- Tables are sparse, only close by pairs
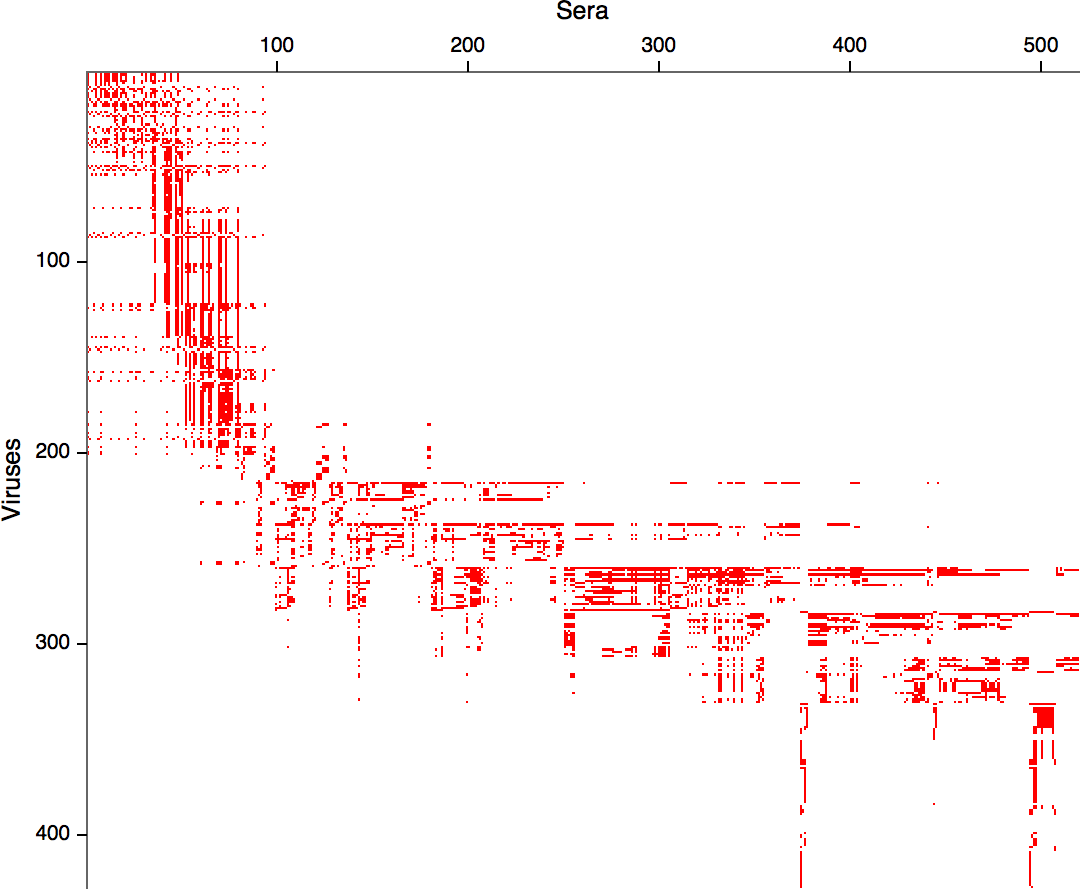
Integrating antigenic and molecular evolution -- ferret serology
 RN et al, PNAS, 2017
RN et al, PNAS, 2017
Accurate prediction remains a challenge!
SARS-CoV-2
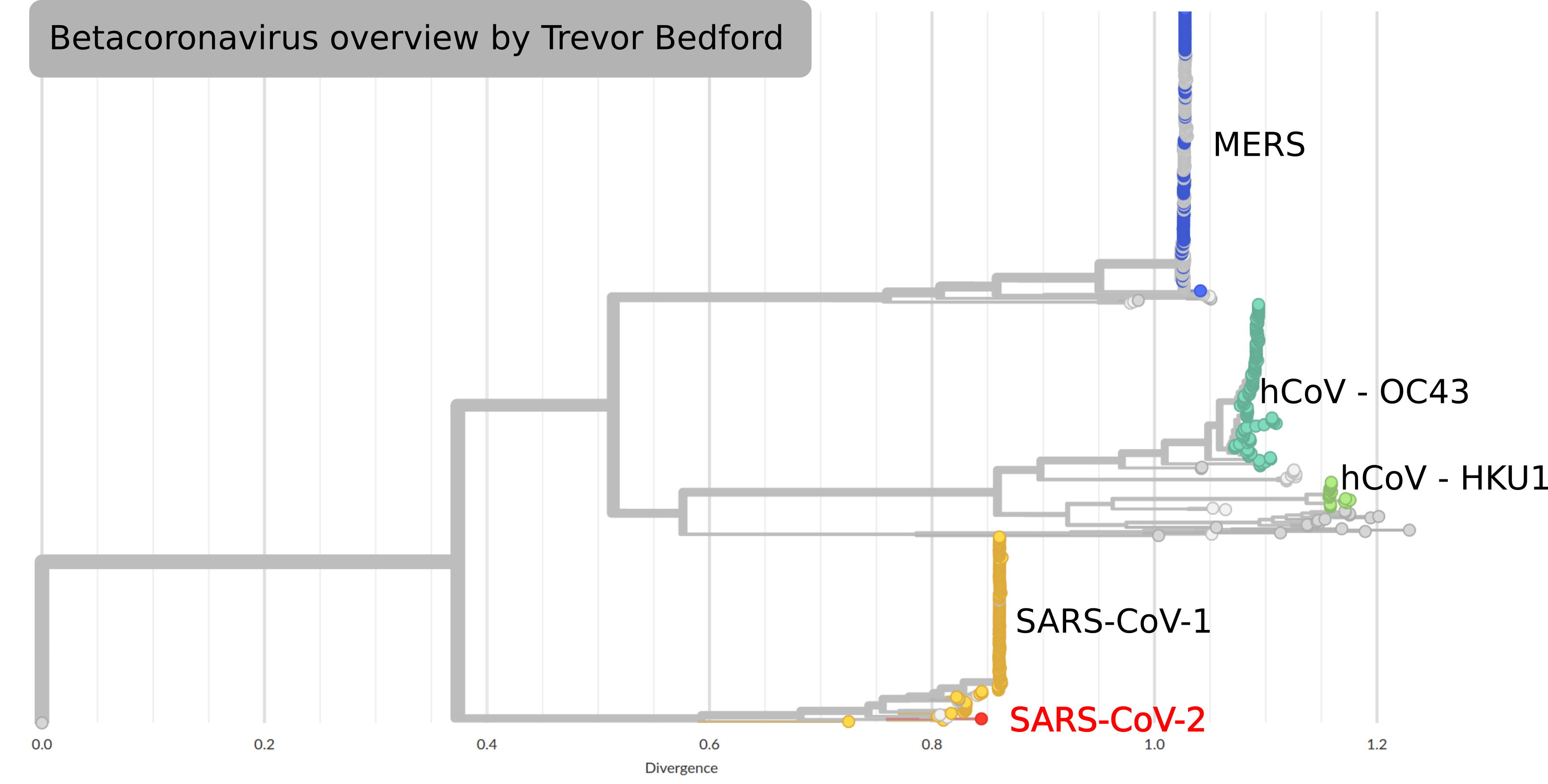 by Trevor Bedford
by Trevor Bedford
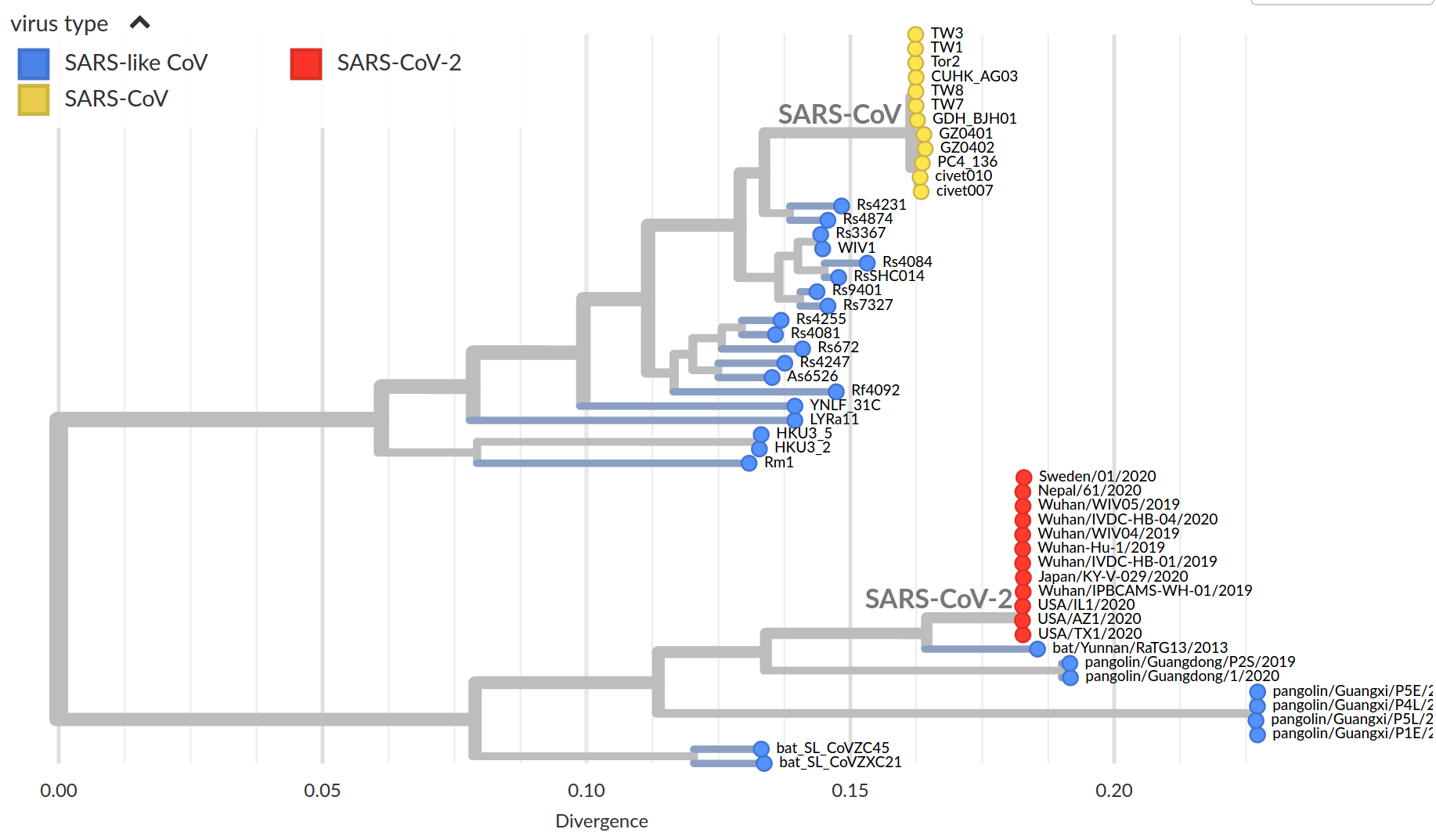 by Trevor Bedford
by Trevor Bedford
Available data on Jan 26 2020
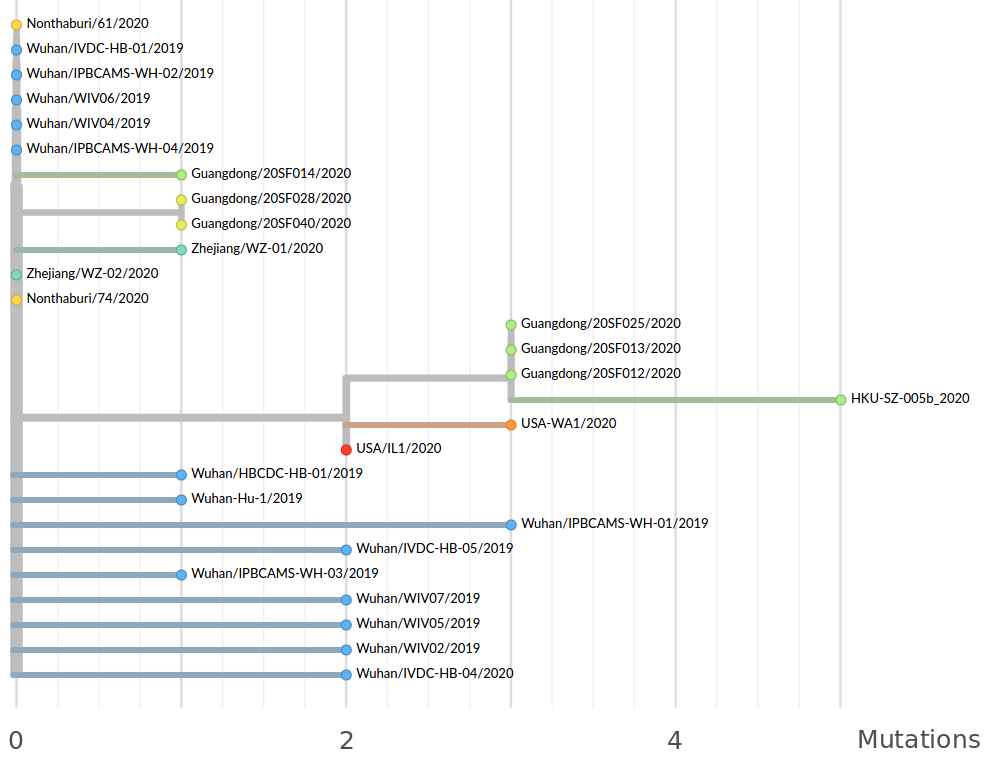
Early genomes differed by only a few mutations, suggesting very recent emergence
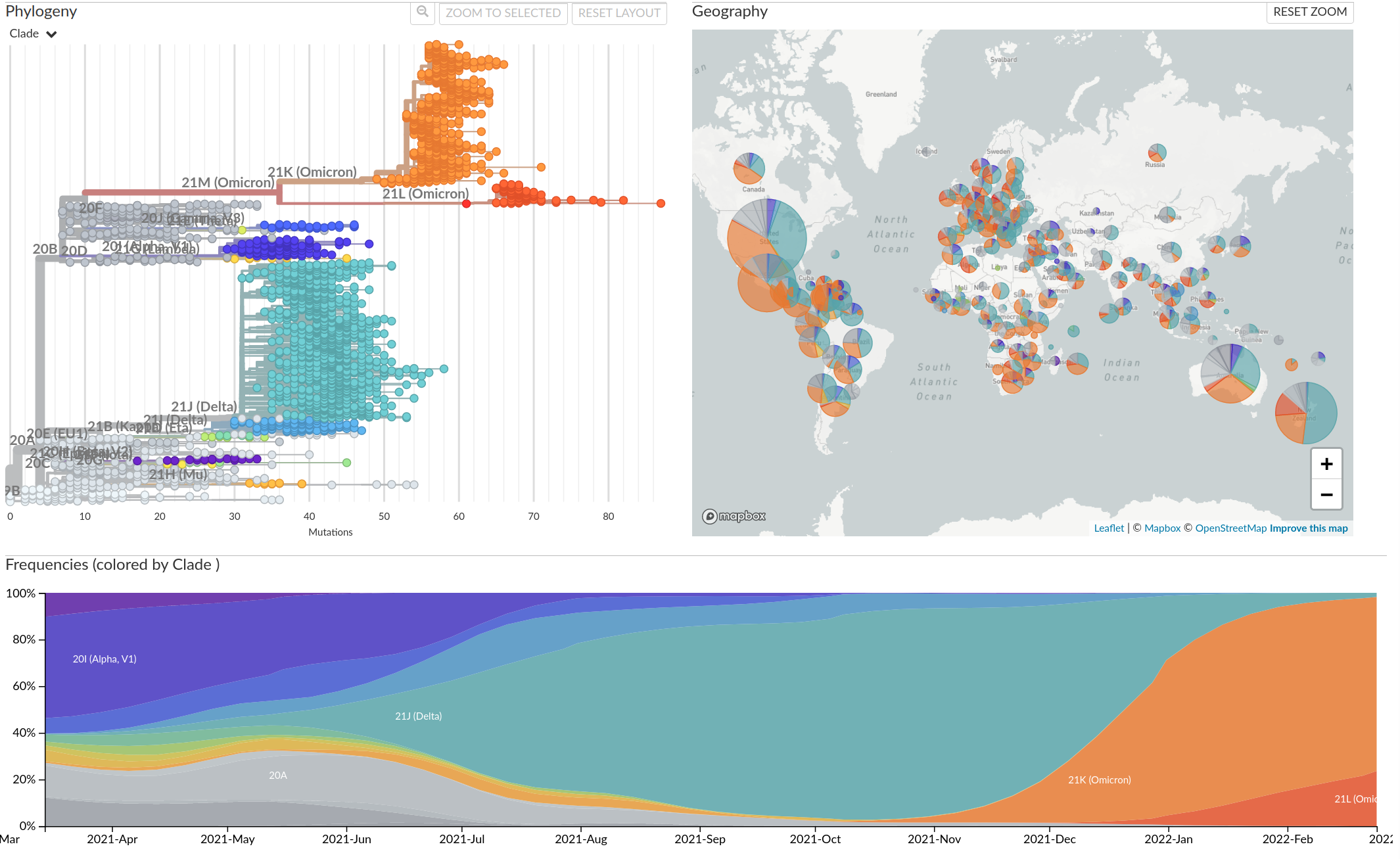
Tracking diversity and spread of SARS-CoV-2 in Nextstrain
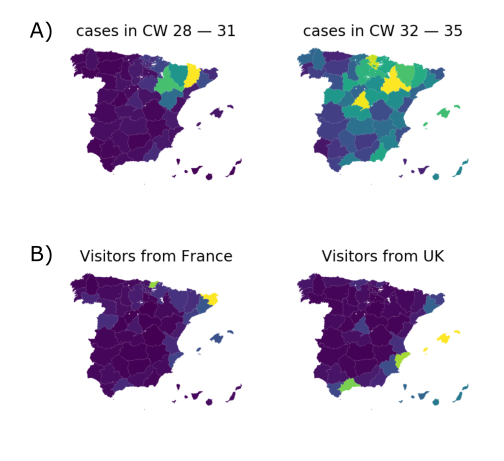
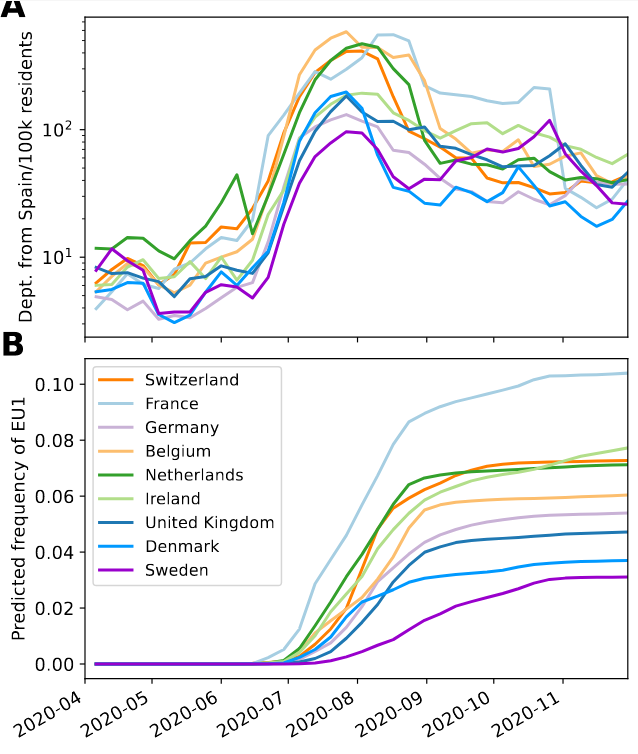
A variant seemed to spread systematically in Summer 2020
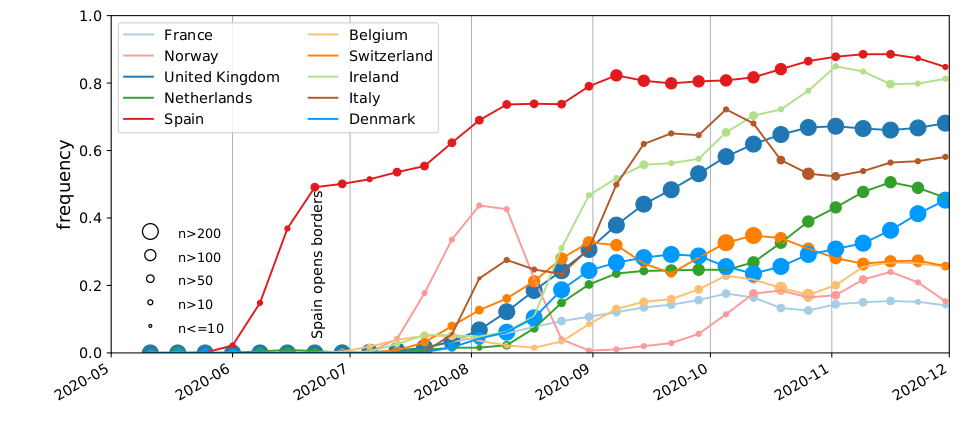
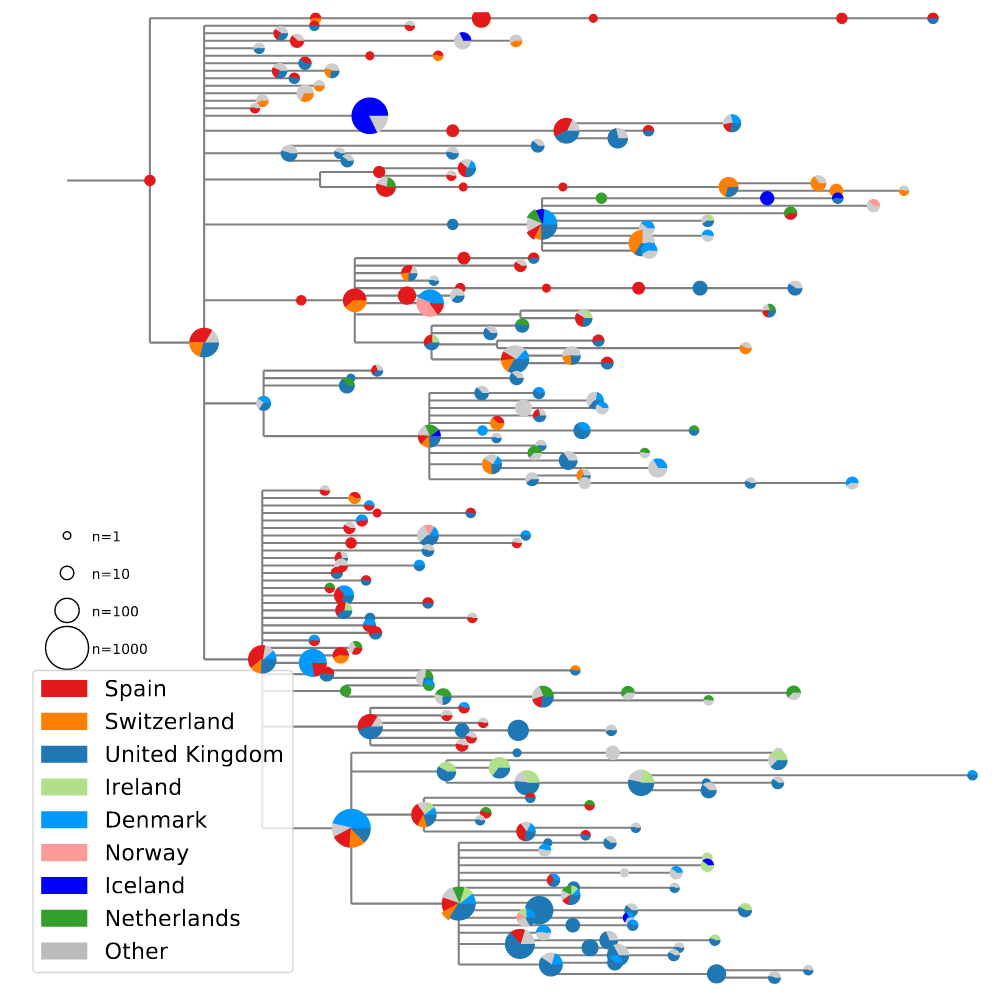 Hodcroft et al
Hodcroft et al
High case numbers in Spain and high travel volume spread the variant
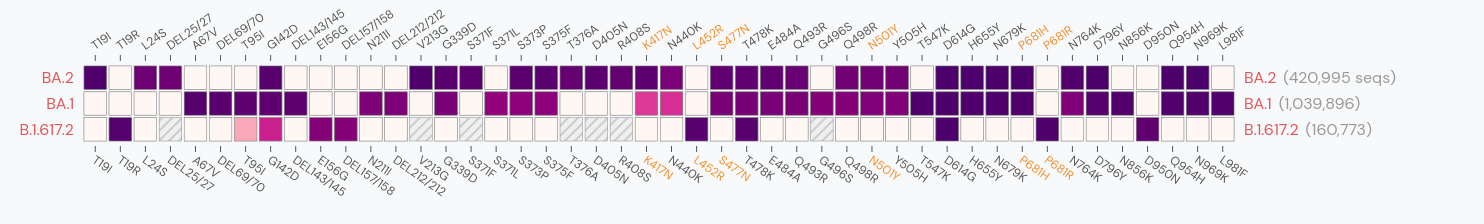
Spike mutations in Omicron
Prediction of SARS-CoV-2 evolution
- Lots of data: around 10 million sequences now
- However: not big data in the traditional sense
- Fitting frequency dynamics to mutations and sequence/variant features.
→ complex epistatic interactions prevent extrapolation to new variants - Using laboratory data to map effects of all possible mutations (deep mutational scanning).
→ immune escape can be assessed from sequence, virulence and fitness not
Too many aspects of SARS-CoV-2 evolution are unknown to make meaningful predictions.
So far, independent variants have dominated successively
Medium term dynamics of SARS-CoV-2 is very uncertain
- Will we start seeing second and third generation variants, as opposed to sister variants?
- Will we the saltatory dynamics with heavily diverged variants continue?
- Will a more diverse immunity landscape slow down future variant dynamics?
- Will waning/antigenic evolution slow down and give rise to annual or even rarer waves?
Going forward, how diverse is SARS-CoV-2 going to be?
Influenza and Theory acknowledgments





- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Pierre Barrat
- Oskar Hallatschek
- All the NICs and WHO CCs that provide influenza sequence data
- The WHO CCs in London and Atlanta for providing titer data
SARS-CoV-2 acknowledgements

.jpg)
- Emma Hodcroft (now in Bern)
- Moira Zuber (Basel)
- Iñaki Comas and Fernando Gonzalez-Candelas, Valencia
- Martina Reichmuth and Christian Althaus (Bern)
- Tanja Stadler, Sarah Nadeau, Tim Vaughan at ETH
- Alberto Hernando and David Matteo at Kido Dynamics
- Jesse Bloom, Katherine Crawford at Fred Hutch
- David Veesler, Alex Walls, Davide Corti, John Bowen at UW
Acknowledgments
Trevor Bedford and his lab -- terrific collaboration since 2014
