Moving targets: Genomic epidemiology tools during a pandemic
Richard Neher
Biozentrum & SIB, University of Basel
slides at neherlab.org/202209_IMMEM.html
From 1'000s to 1'000'000s of samples
From a few labs to hundreds of contributing labs
Our favorite fancy tools and models couldn't handle it
- Sample size: could only use a tiny fraction of data
- Slow: Analyses that take weeks are incompatible with actionable insights.
- Complex: Link between inferences and data signatures too indirect.
Needed tools to make sense of an avalanche of data
- Fast: immediate results/summaries
- Robust: data quality varies, sampling is biased. Complex models go wrong in weird ways when assumptions are violated.
- Simple: anything to complex can't be interpreted reliably.
Many smart people made amazing tools with open data
- cov-spectrum: interactive interface to query any mutation/lineage
- Usher: find related viruses in giant tree, assign pango-lineages
- outbreak.info: variant/mutation stats
- cov2tree.org: giant SC2 phylogenies
- PANGO lineages: granular lineages for tracking
Sequence analysis and interpretation are challenging
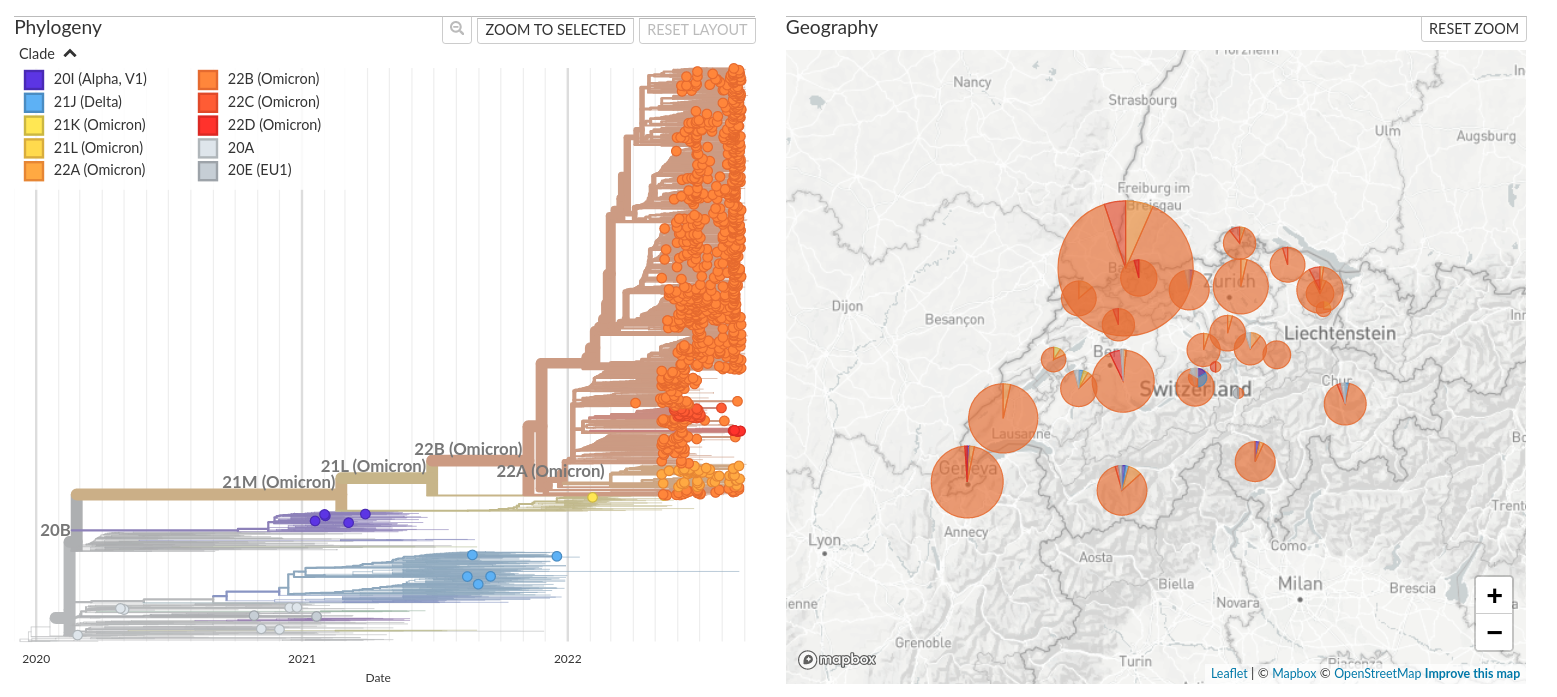
Nextstrain's focus: enable teams to make sense of their data
Workflows to analyze custom data + background
- Hierarchical sampling: global, country, division.
- Hosting via Nextstrain groups
- Aimed at completion within hours
- Data sharing restriction made this more difficult than it should have been
- No experience necessary
- QC: avoid releasing bad data
- Clades, lineages, mutations
- Private
Nextclade
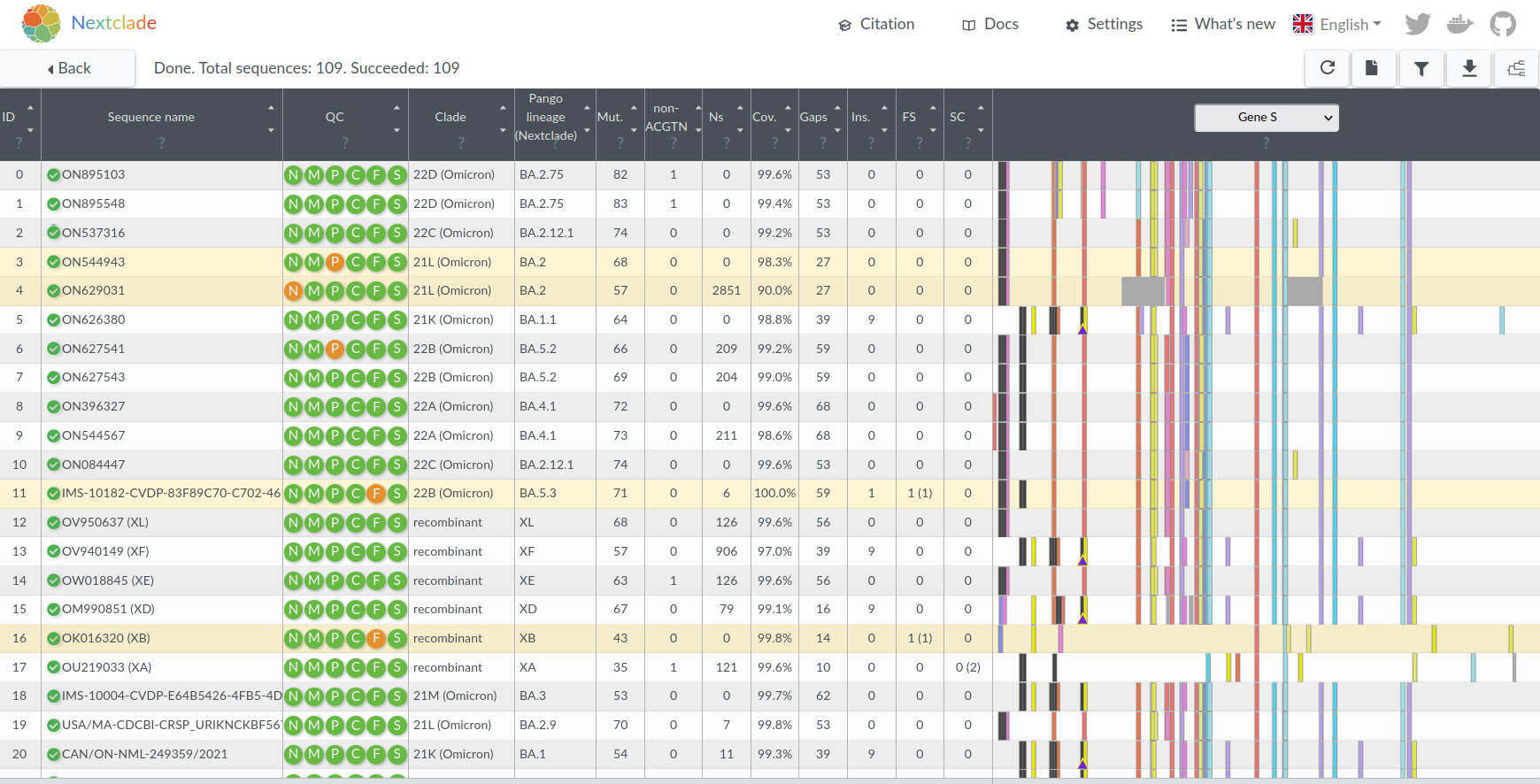
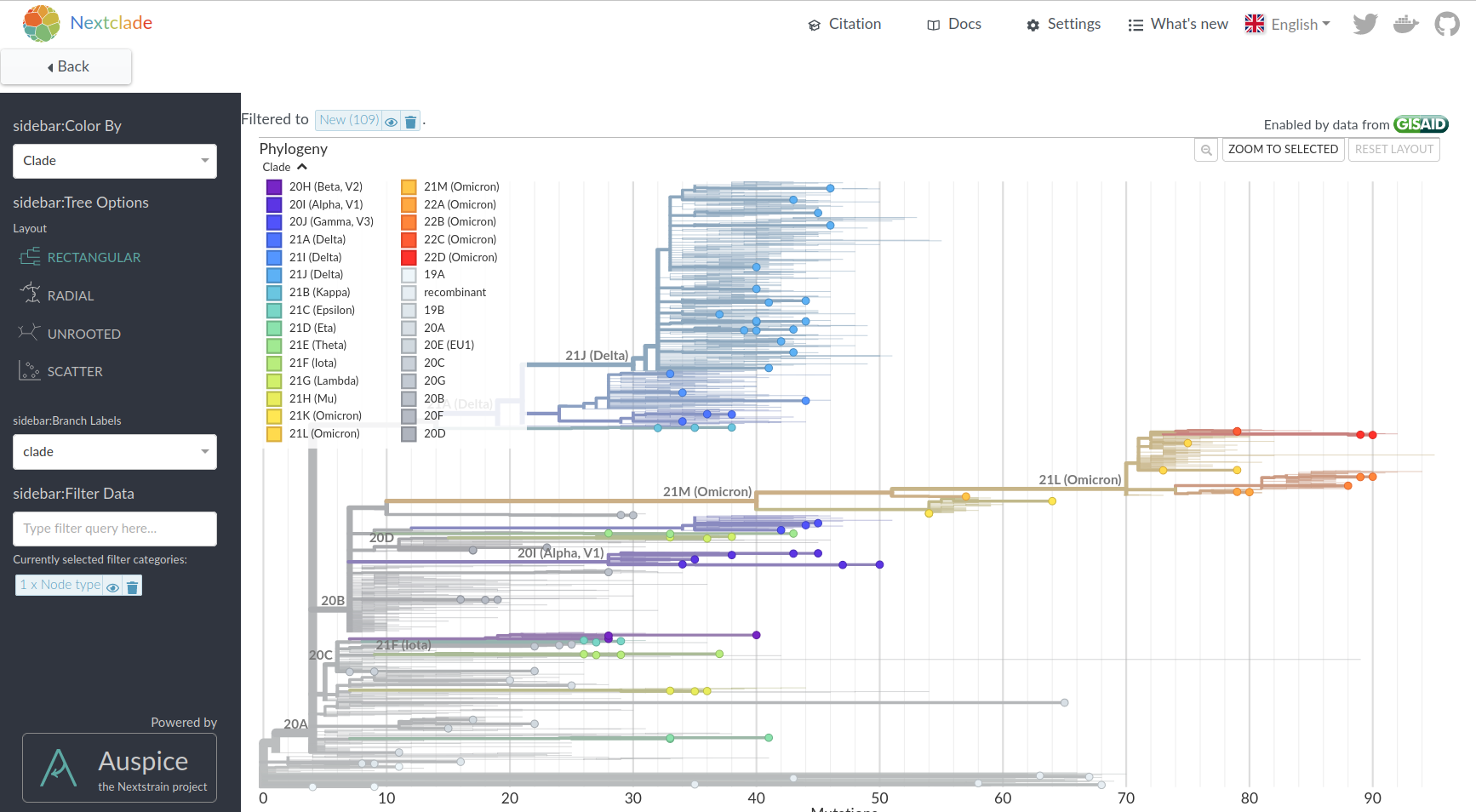
Nextclade Web and CLI
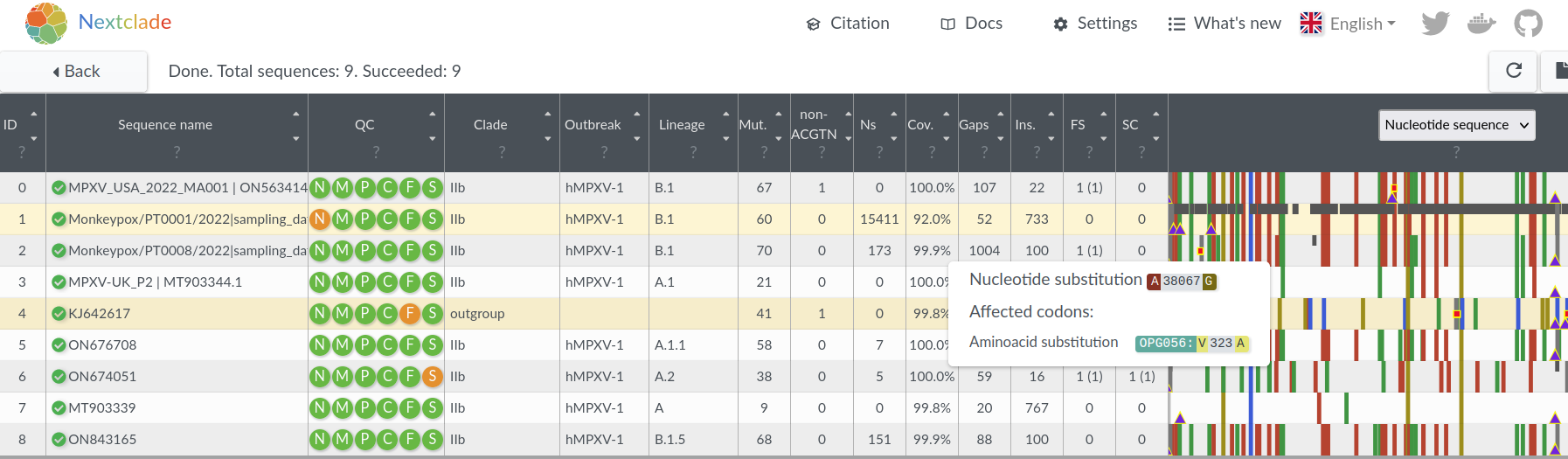
- Aligns, translates, classifies a SARS-CoV-2 genome in 20ms
- Now used for QC and filtering in many Nextstrain workflows
- QC and mutations calls used for many downstream analysis
All open sequences annotated and aligned, updated daily:
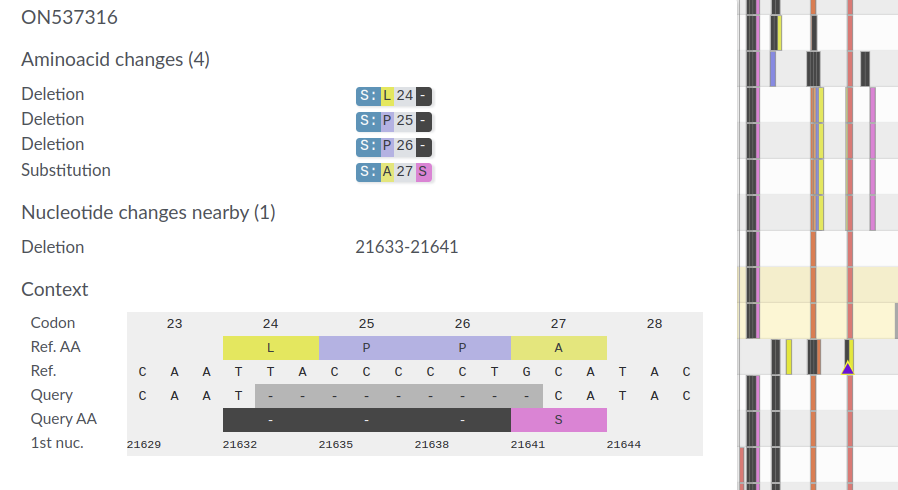
May 2022: Monkeypoxvirus
- SARS-CoV-2 is a big RNA virus (30kb), the MPXV genome is 200kb long.
- Repeats, low complexity regions, etc...
- Can we still align this in the browser?
Acknowledgements


Ivan Aksamentov
Cornelius Roemer
Cornelius Roemer

Data are contributed by scientists from all over the world and curated by Genbank or GISAID

