Virus evolution and the spread of infectious disease
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/202211_Ringvorlesung.html
Viruses
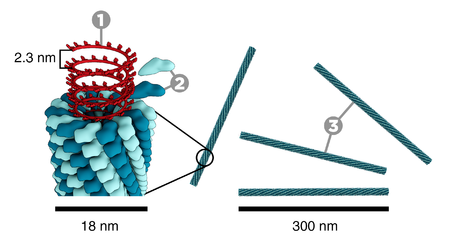 tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
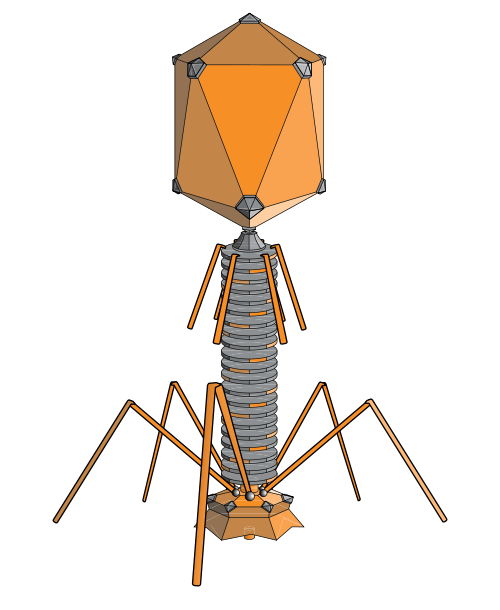
bacteria phage (adenosine, wikipedia)
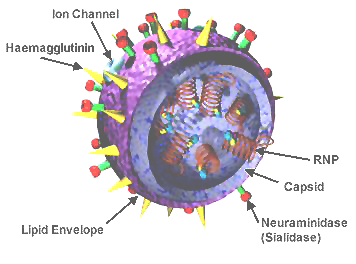
influenza virus wikipedia

human immunodeficiency virus wikipedia
- rely on host to replicate
- little more than genome + capsid
- genomes typically 5-200k bases (+exceptions)
- most abundant organisms on earth $\sim 10^{31}$
Lifecycle of animal viruses
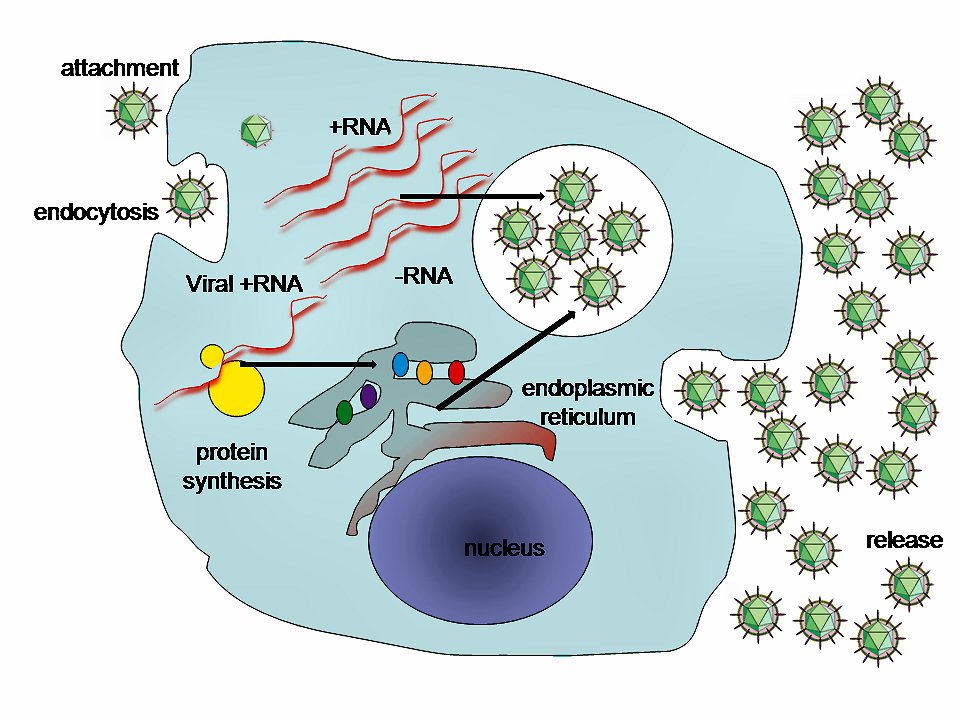 By GrahamColm at English Wikipedia
By GrahamColm at English Wikipedia
Development of sequencing technologies
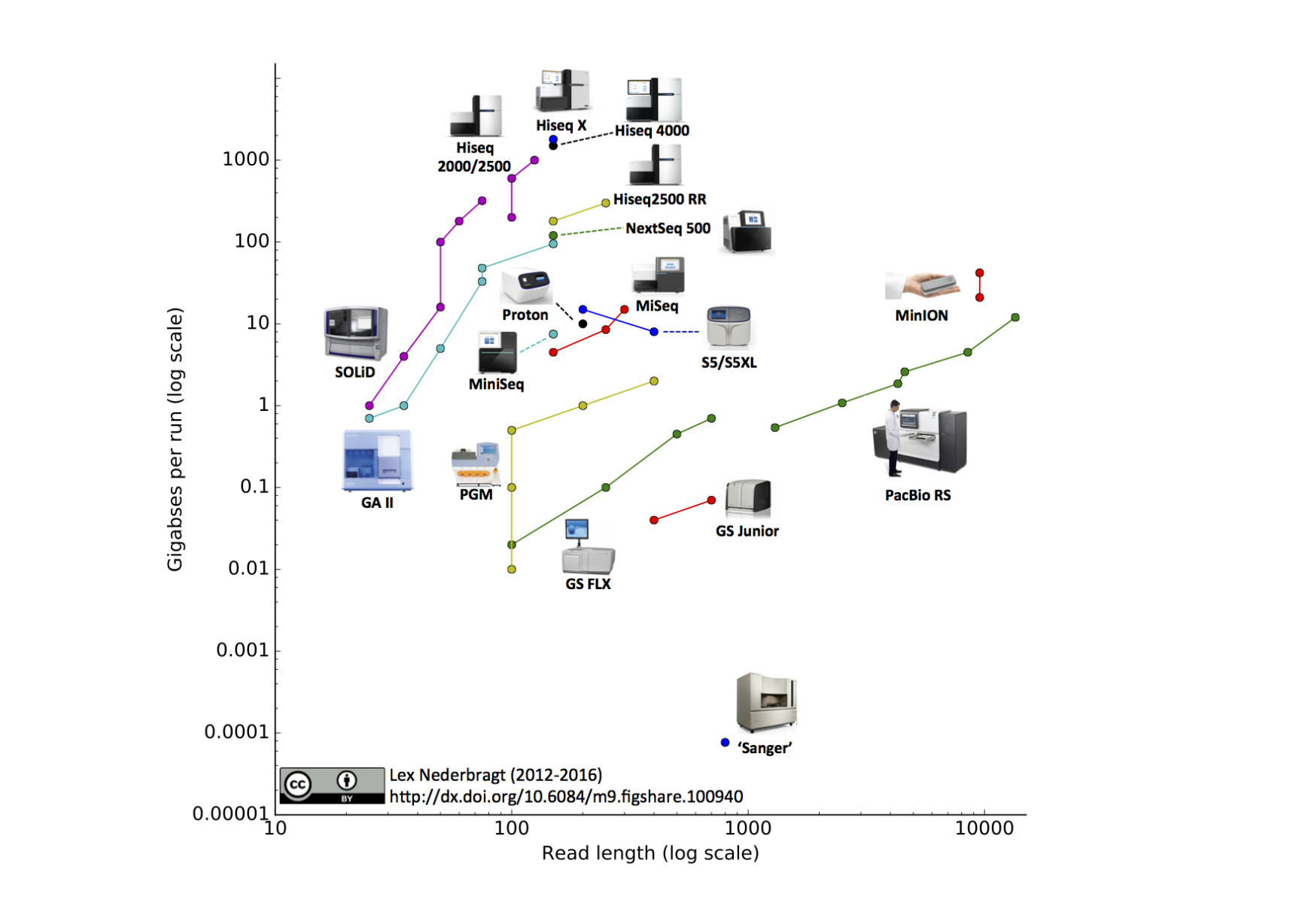
We can now sequence...
- thousands of bacterial isolates
- thousands of single cells
- populations of viruses, bacteria or flies
- diverse ecosystems
Phylogenetic analysis of viral sequences


RNA viruses have a high mutation rate. New mutations arise every few weeks.
Some viruses evolve a million times faster than animals
Animal haemoglobin

HIV protein

Evolution of HIV
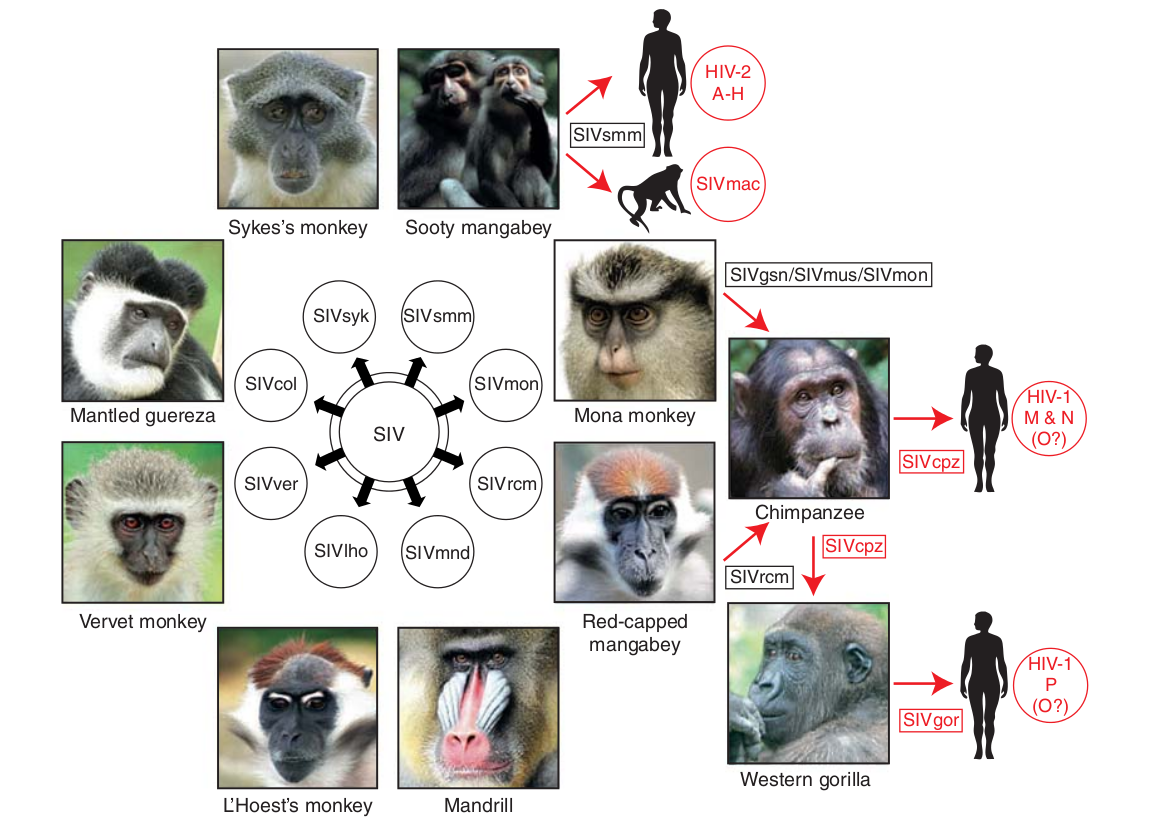
- Chimp → human transmission around 1900 gave rise to HIV-1 group M
- ~100 million infected people since
- subtypes differ at 10-20% of their genome
- HIV-1 evolves ~0.1% per year
HIV infection
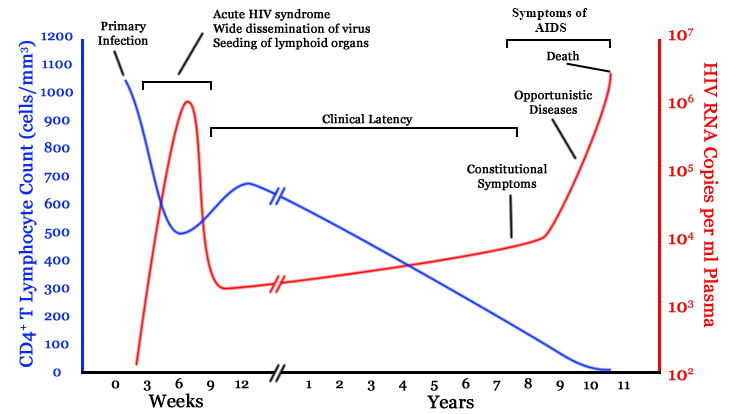

- $10^8$ cells are infected every day
- the virus repeatedly escapes immune recognition
- integrates into T-cells as
latent provirus
{kind=link}
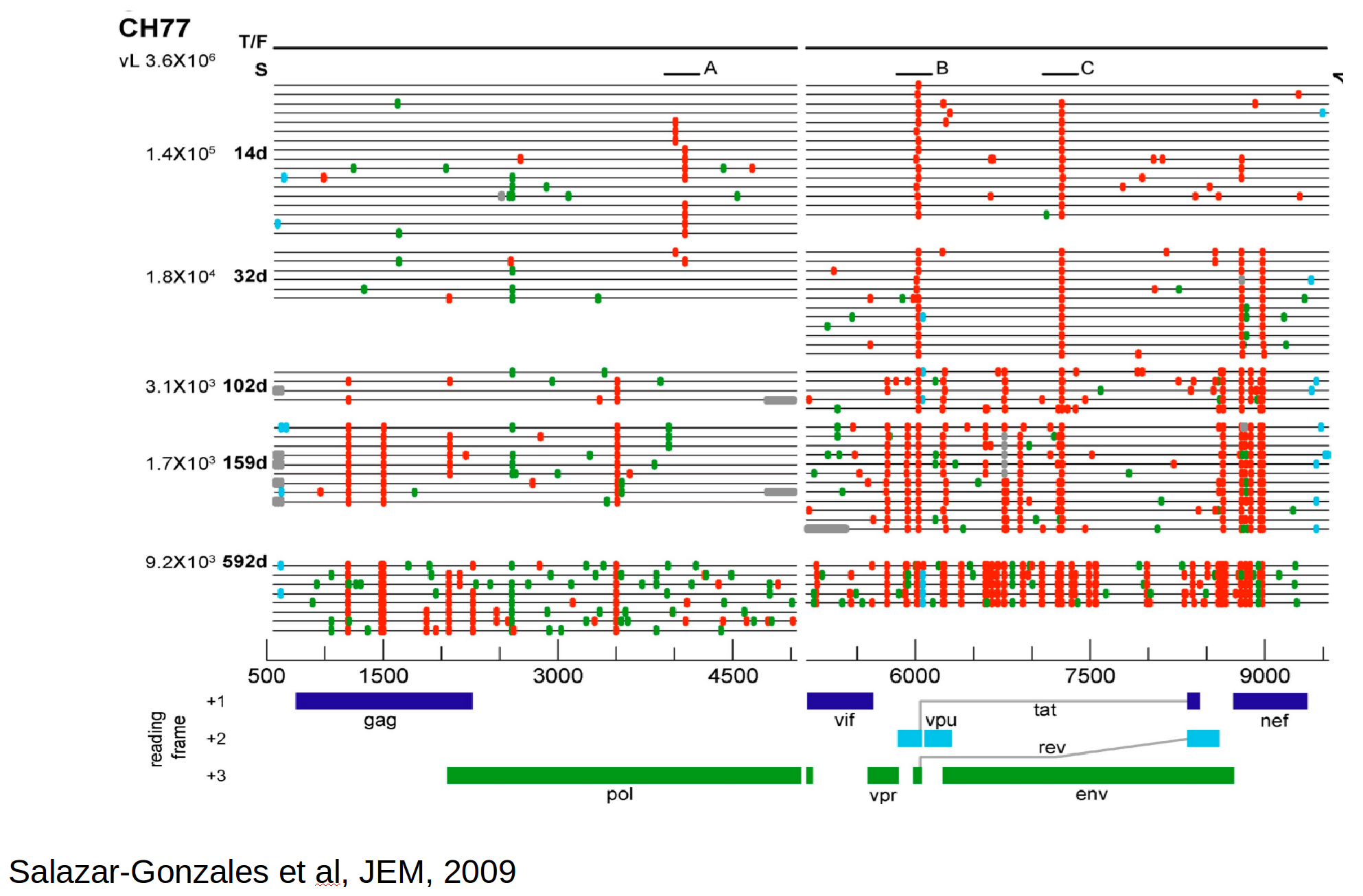
HIV-1 evolution within one individual

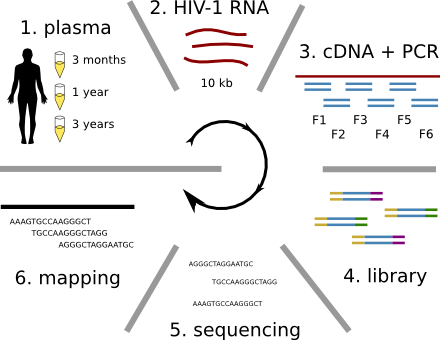

Immune escape in early HIV infection
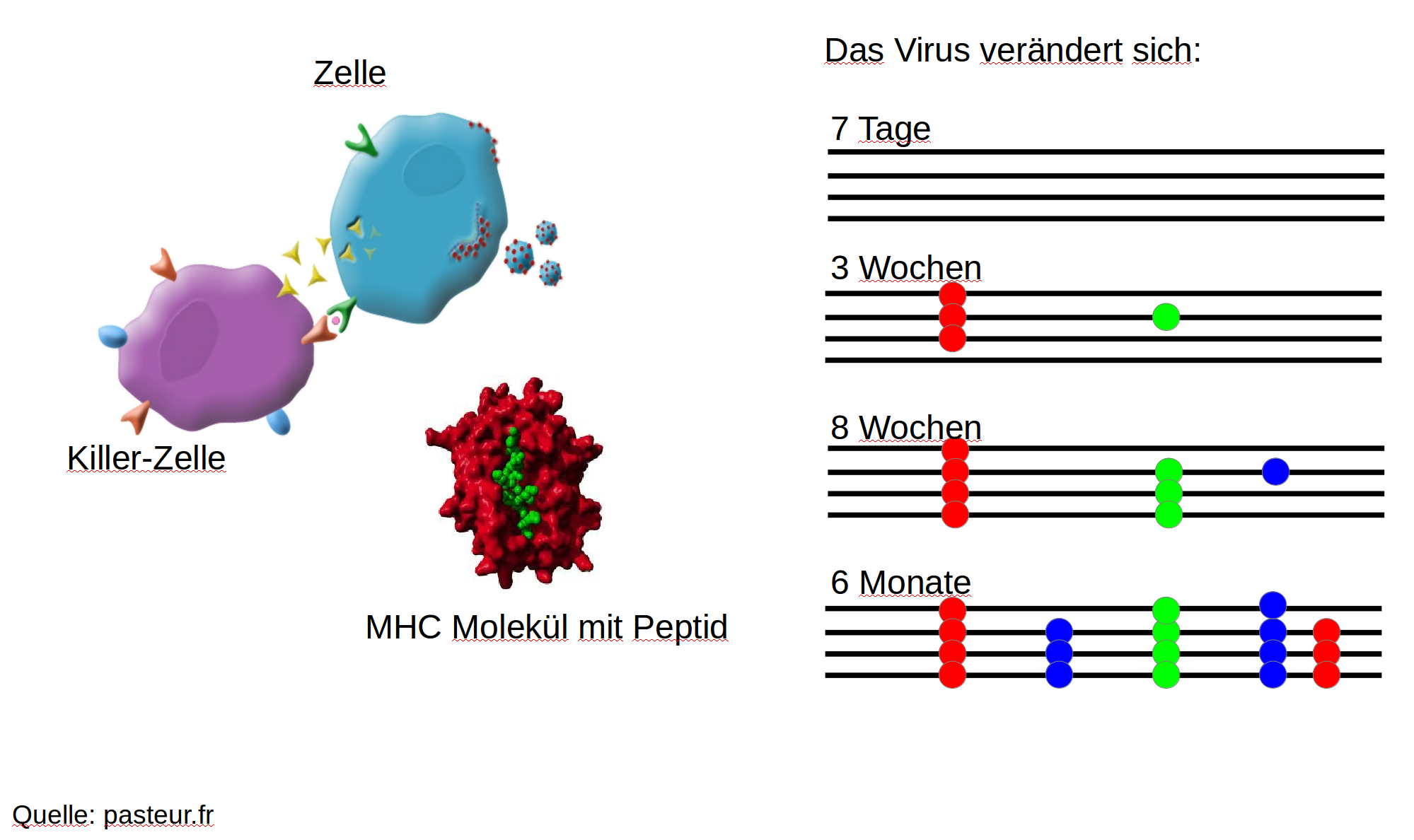
Immune escape in early HIV infection
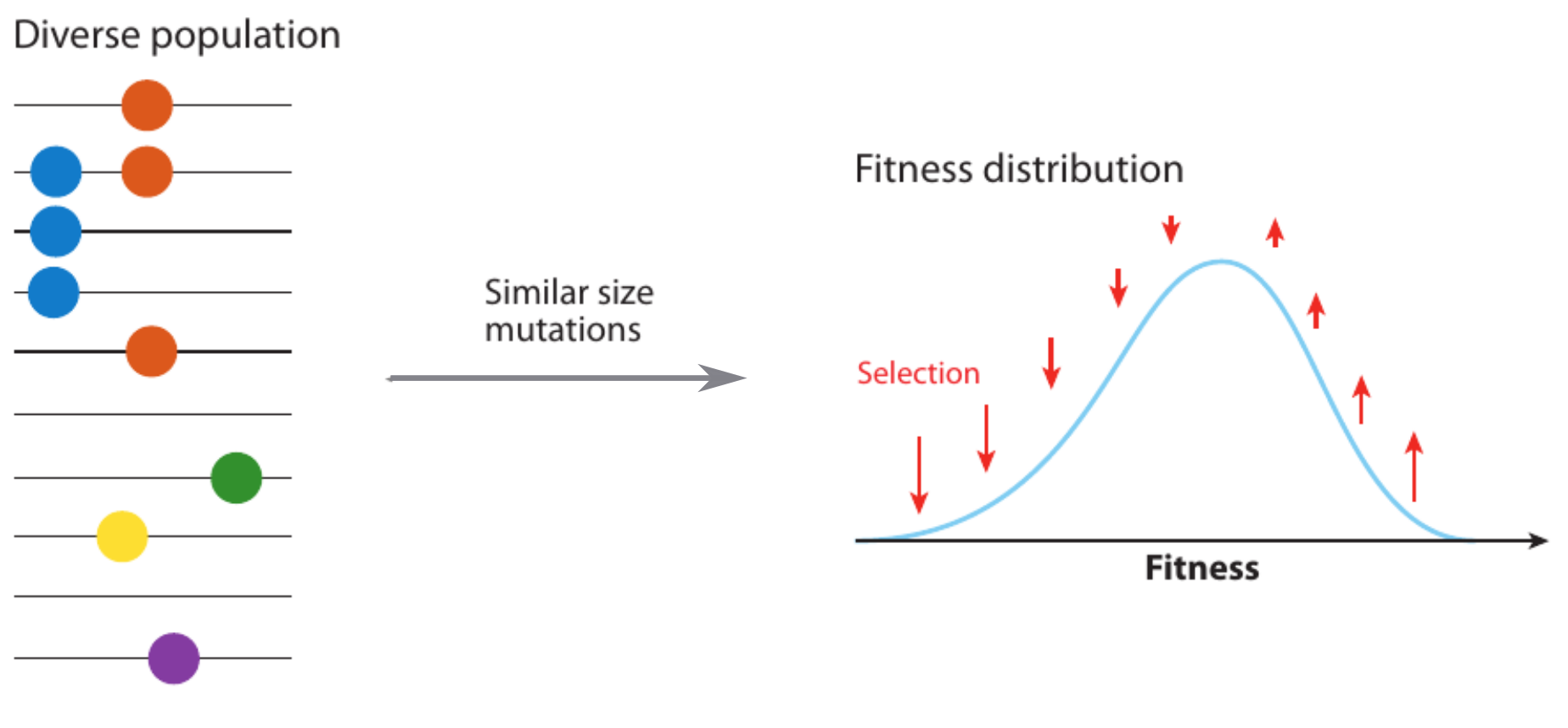
Selective sweeps
- Viruses carrying a beneficial mutation have more offspring: on average $1+s$ instead of $1$
- $s$ is called selection coefficient
- Fraction $x$ of viruses carrying the mutation changes as $$x(t+1) = \frac{(1+s)x(t)}{(1+s)x(t) + (1-x(t))}$$
- In continuous time → logistic differential equation: $$\frac{dx}{dt} = sx(1-x) \Rightarrow x(t) = \frac{e^{s(t-t_0)}}{1+ e^{s(t-t_0)}}$$
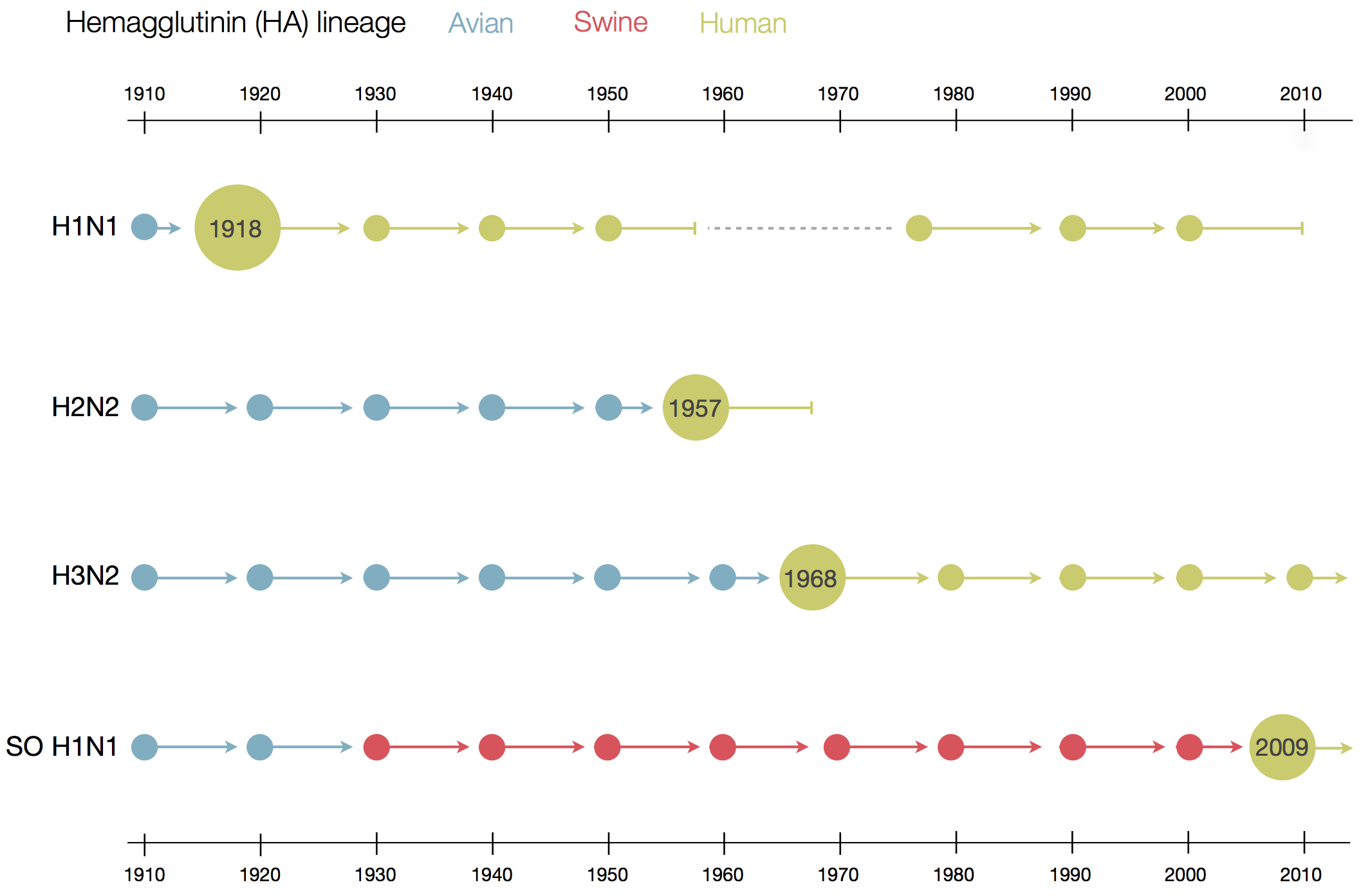
Human Influenza A viruses
Weekly numbers of positive influenza tests in the US by subtype
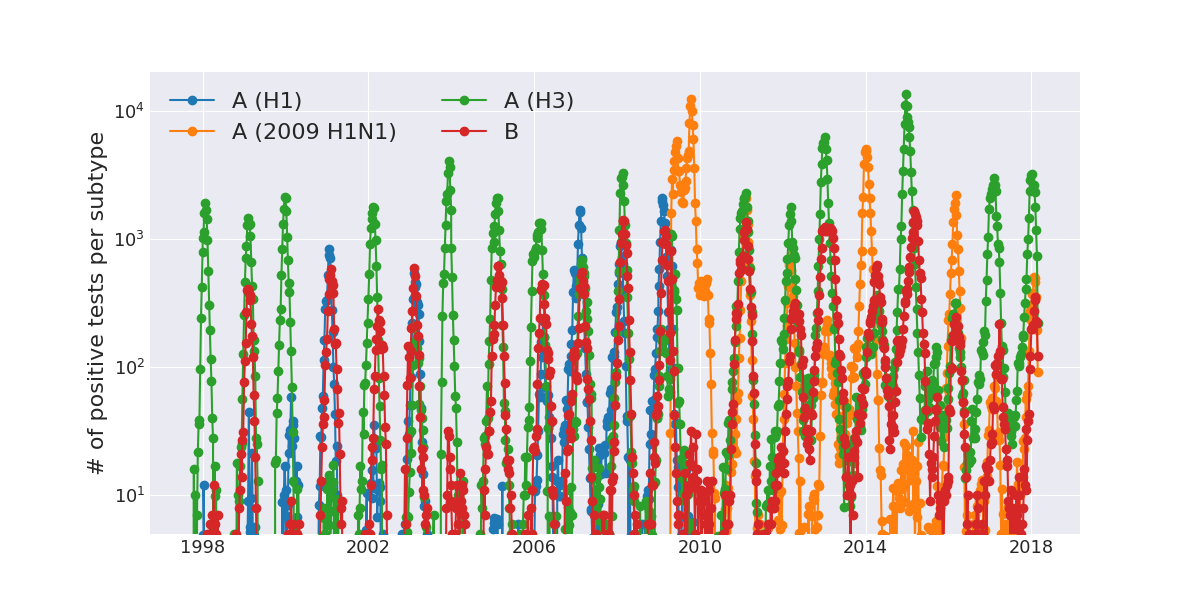 Data by the US CDC
Data by the US CDC
Influenza virus
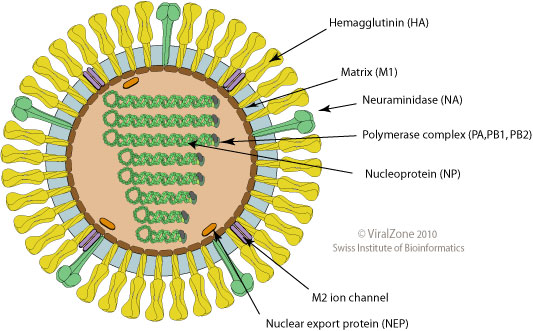
- Surface proteins hemagglutinin (HA) and neuraminidase (NA)
- Influenza A virus
- Common in birds and mammals
- Many different subtypes defined by surface proteins
- H3N2, H1N1, H7N9, H5N1
- Influenza B virus
- infects mainly humans
- two lineages that split 30-40y ago
- B/Victoria vs B/Yamagata
nextflu.org
joint work with Trevor Bedford & his lab
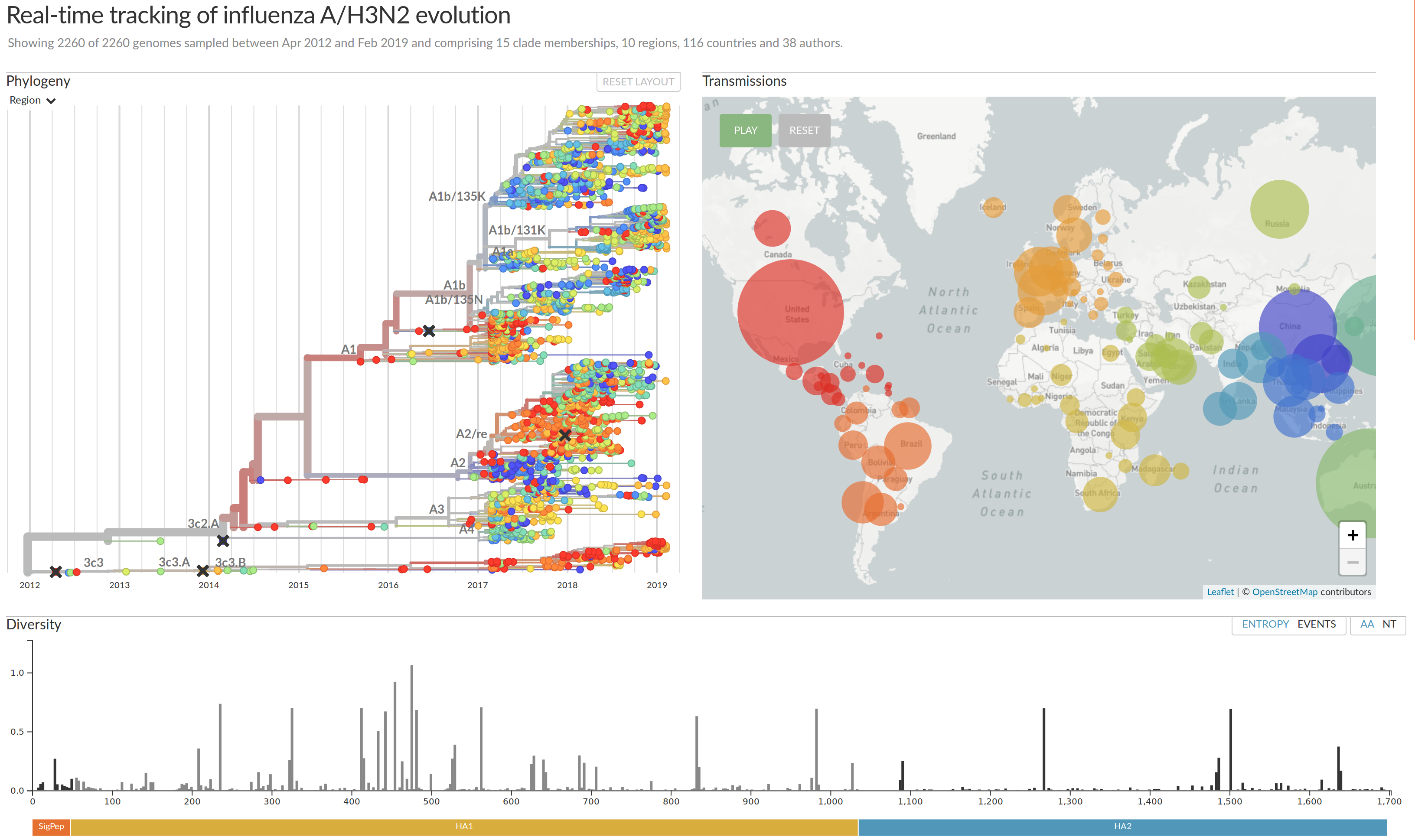
nextstrain.org
joint work with Trevor Bedford & his lab
Clonal interference and traveling waves
Typical tree
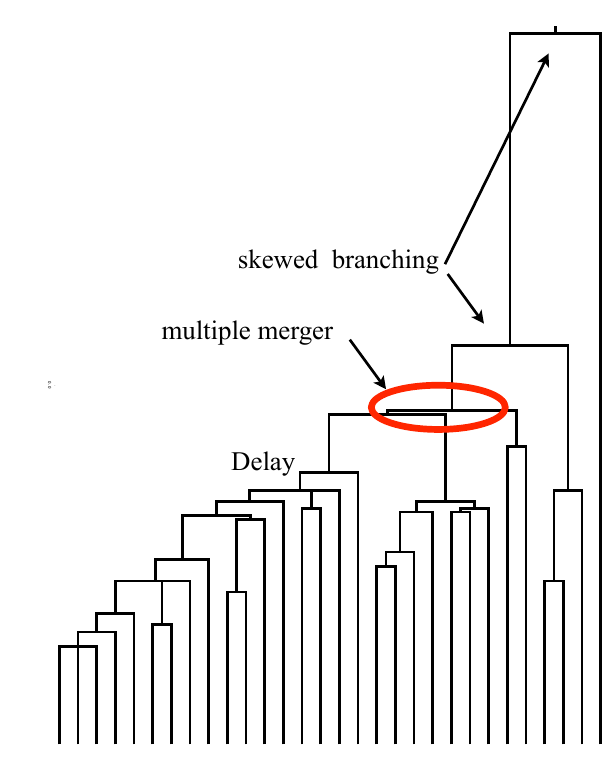
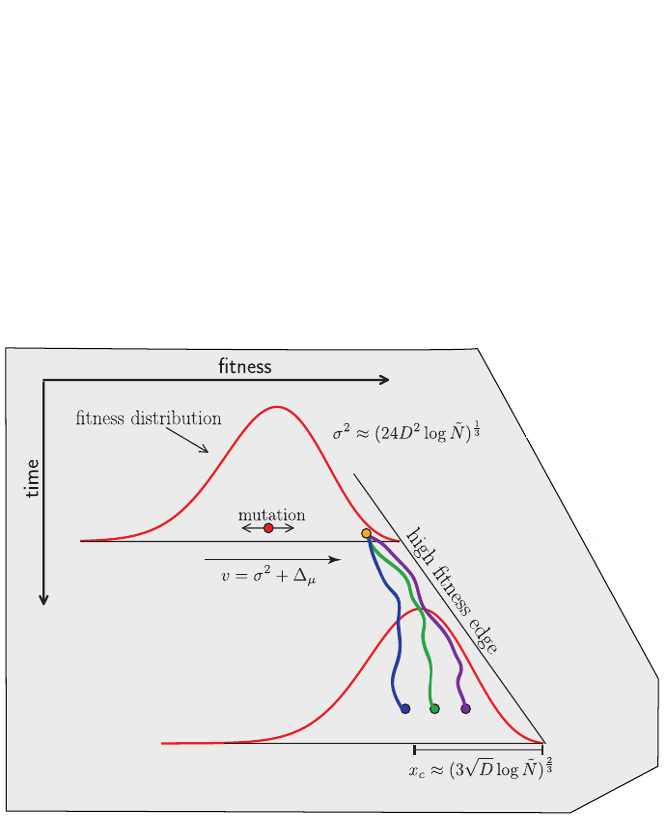
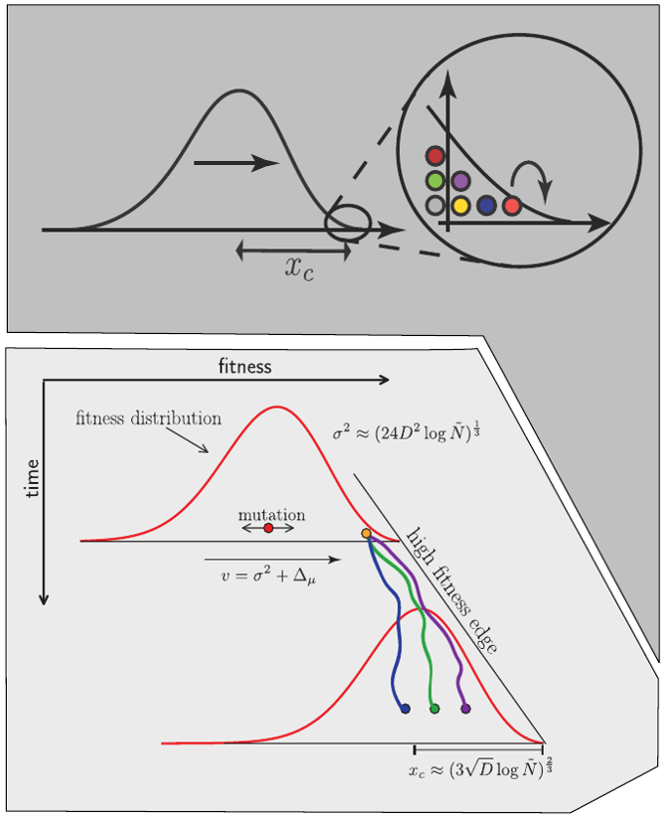
Bolthausen-Sznitman Coalescent
Bursts in a tree ↔ high fitness genotypes
Can we read fitness of a tree?
Predicting evolution
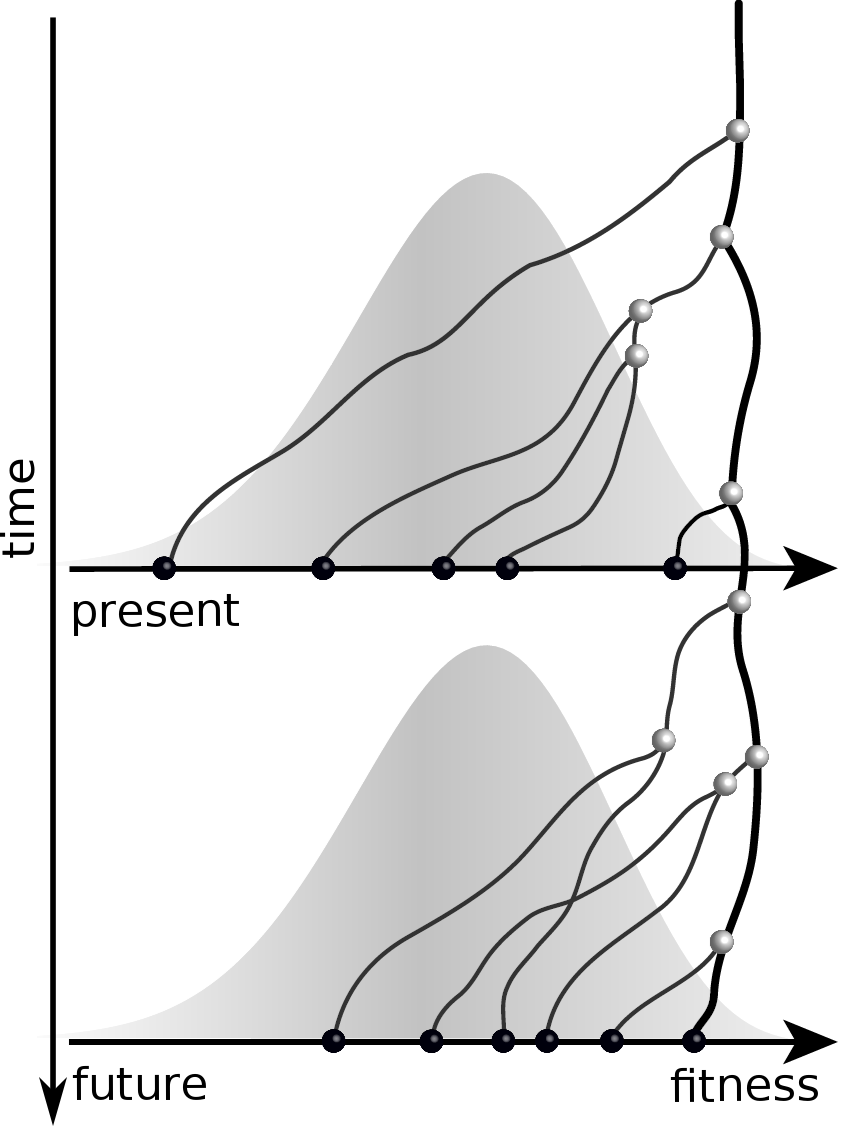
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
Validation on simulated data
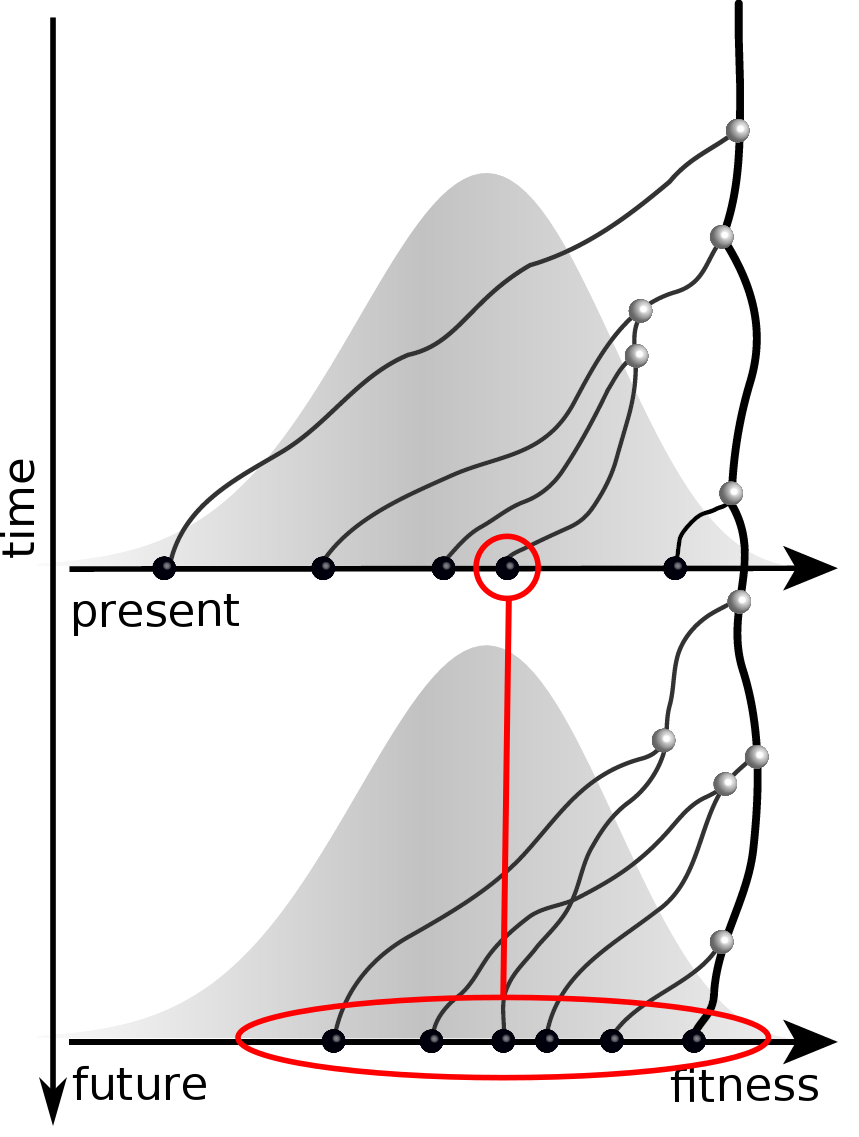
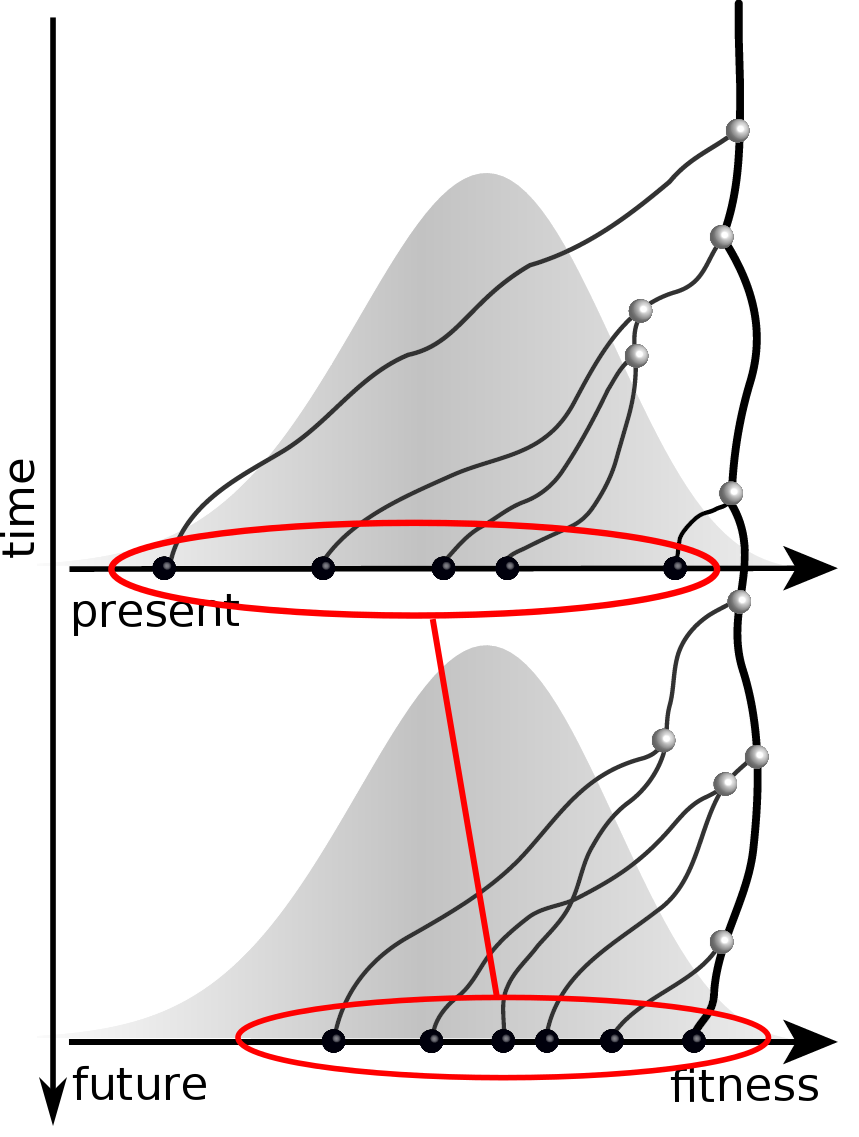
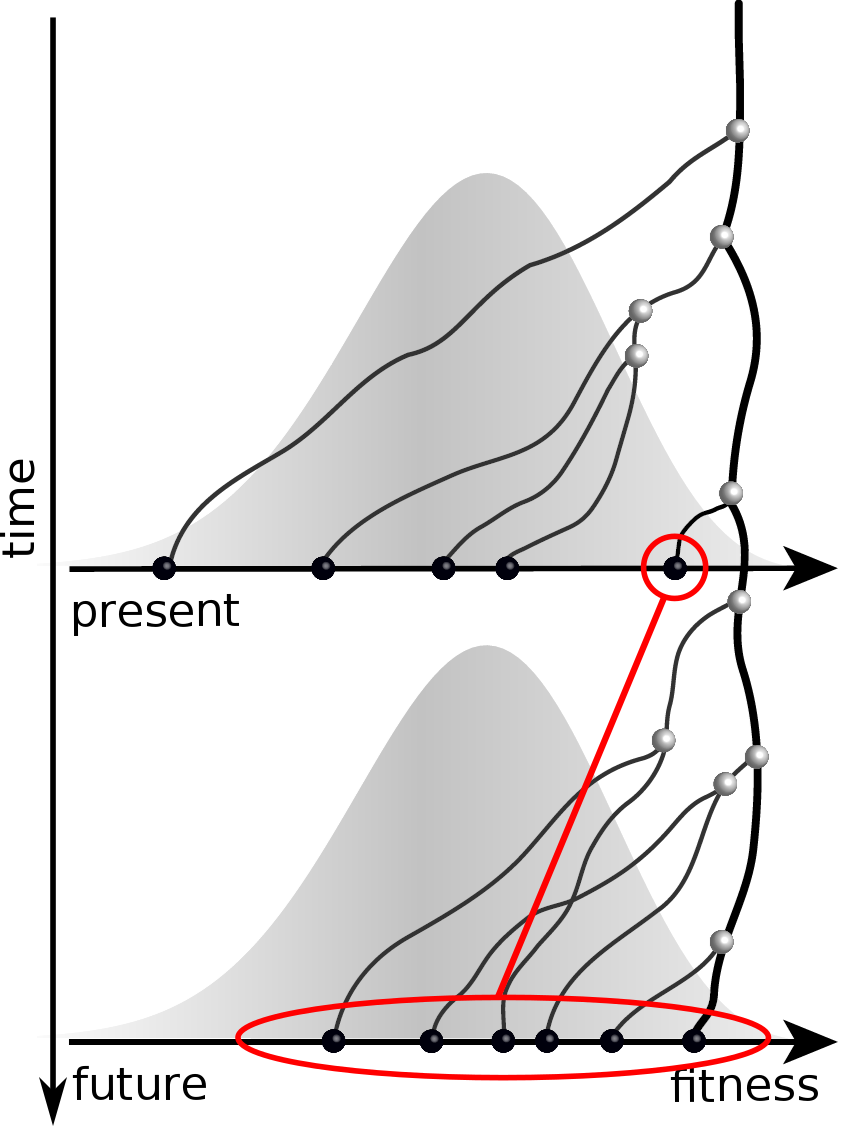
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
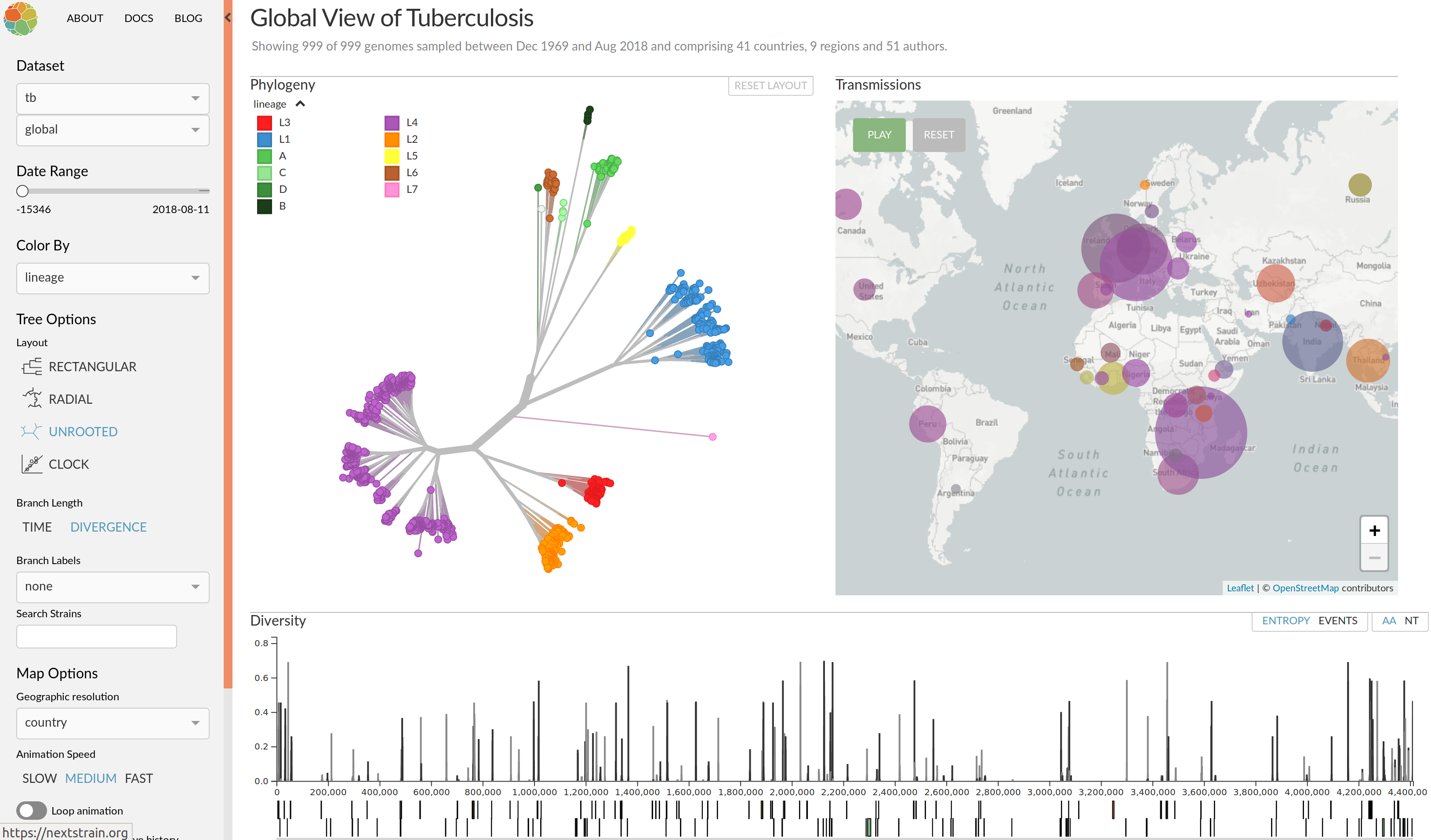
Real-time tracking of SARS-CoV-2
- thousands of new sequences every day
- more than >14M sequences right now
- comprehensive analysis require hours to days to complete
→ requires continuous analysis and easy dissemination
→ interpretable and intuitive visualization
nextstrain.org
joint project with Trevor Bedford & his lab
Emergence and dominance of SARS-CoV-2 variants
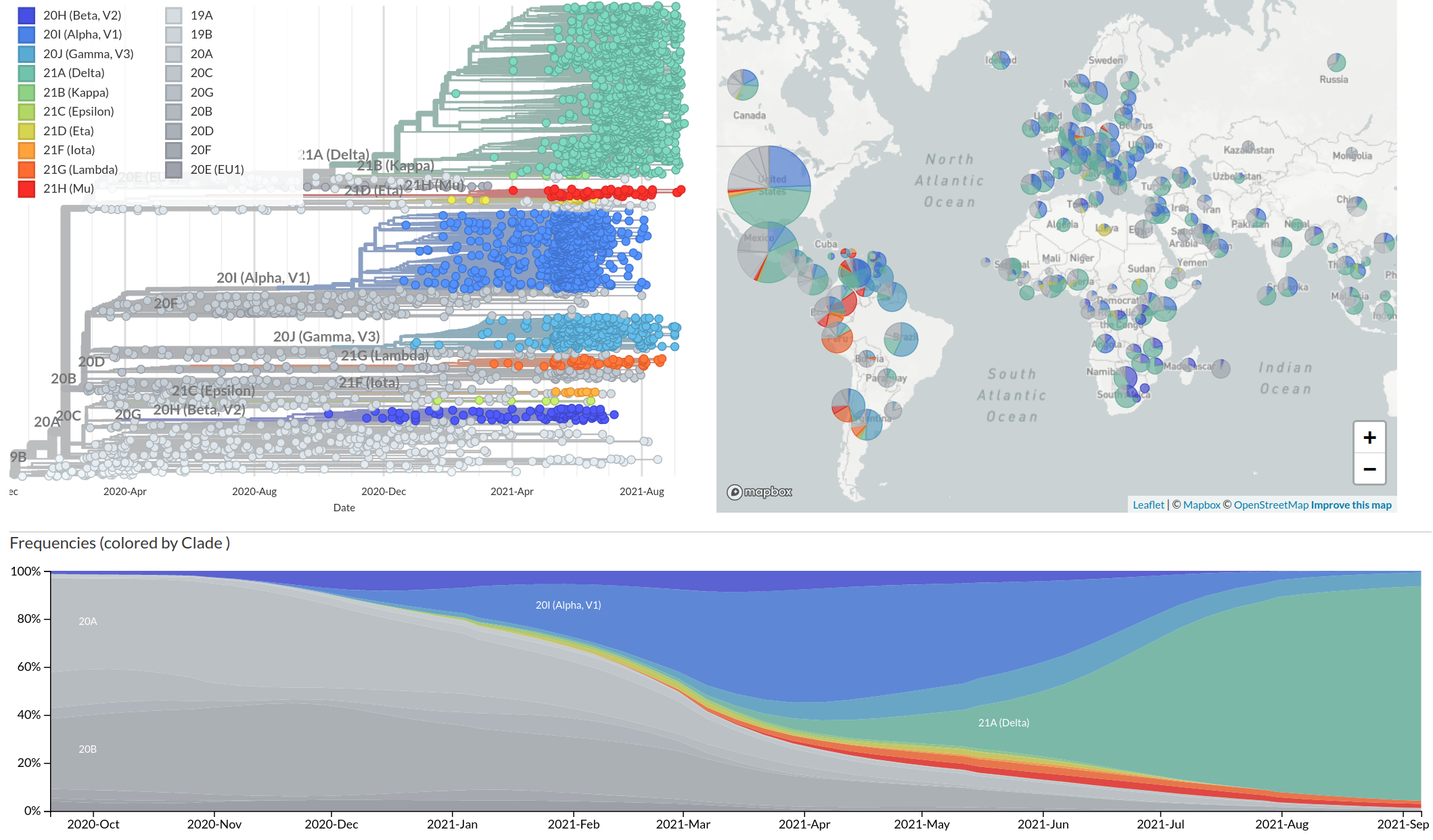
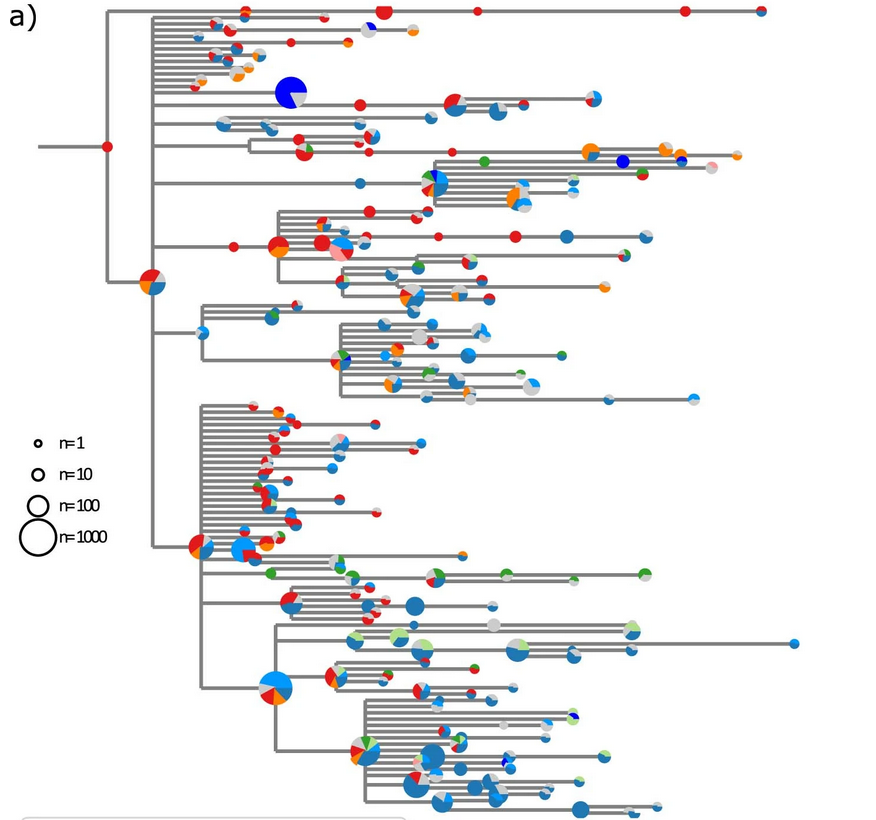
SARS-CoV-2 variants can become dominant without advantage
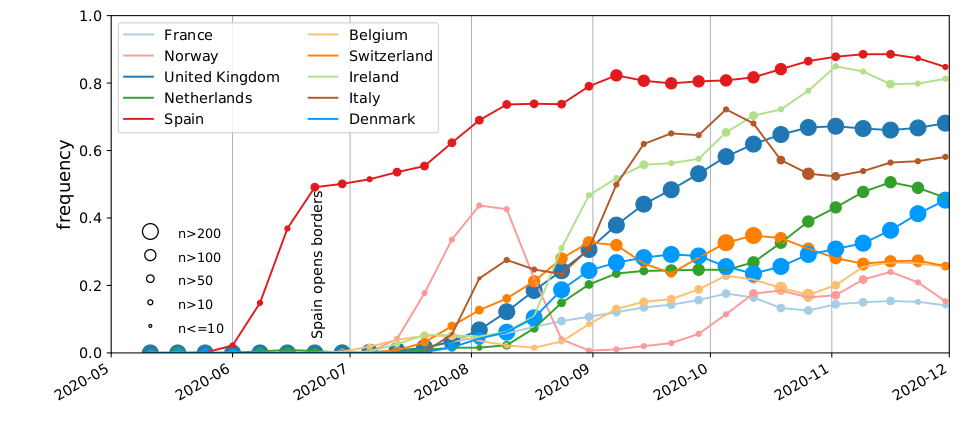
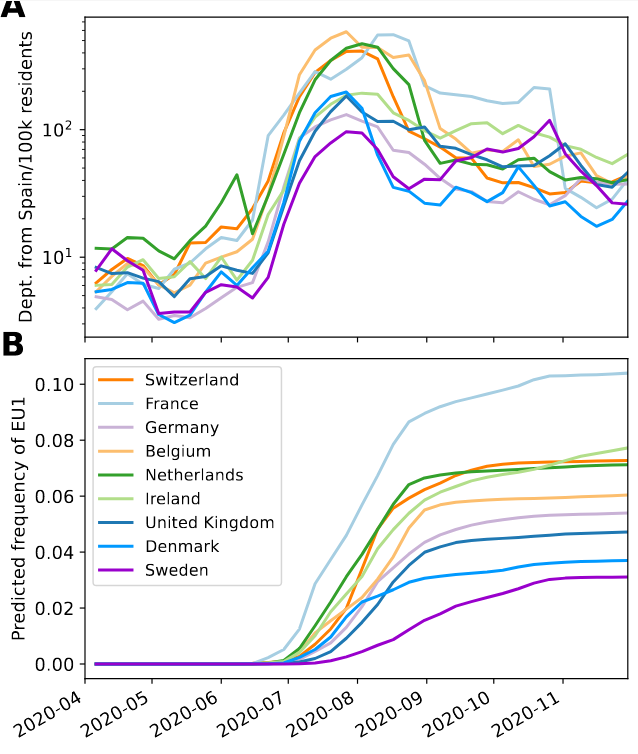
Spanish EU1 diversity was mirrored across Europe

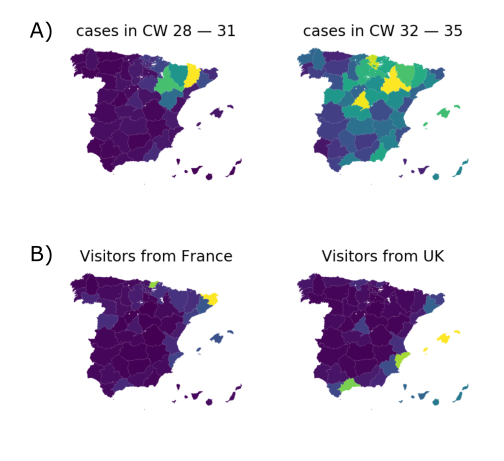
High case numbers in Spain and high travel volume spread the variant
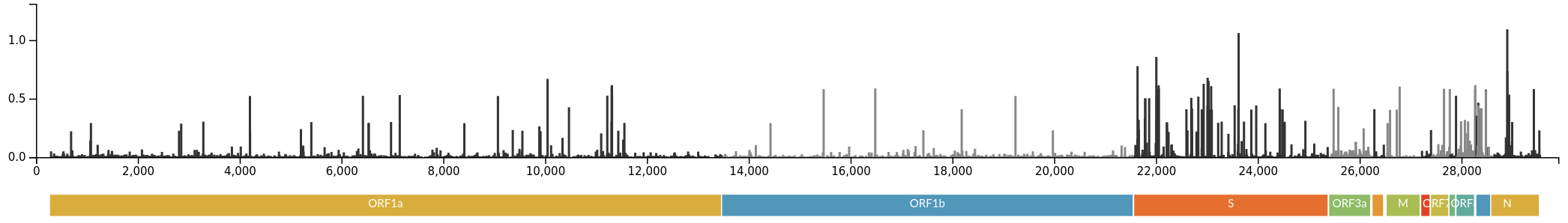
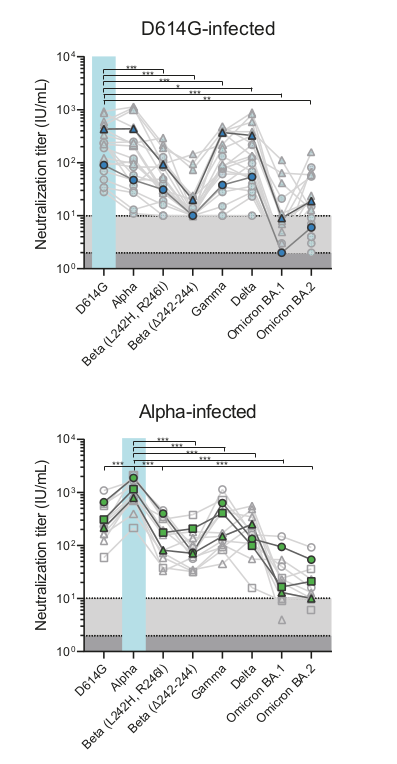
Successful variants are characterized by many mutations in S1
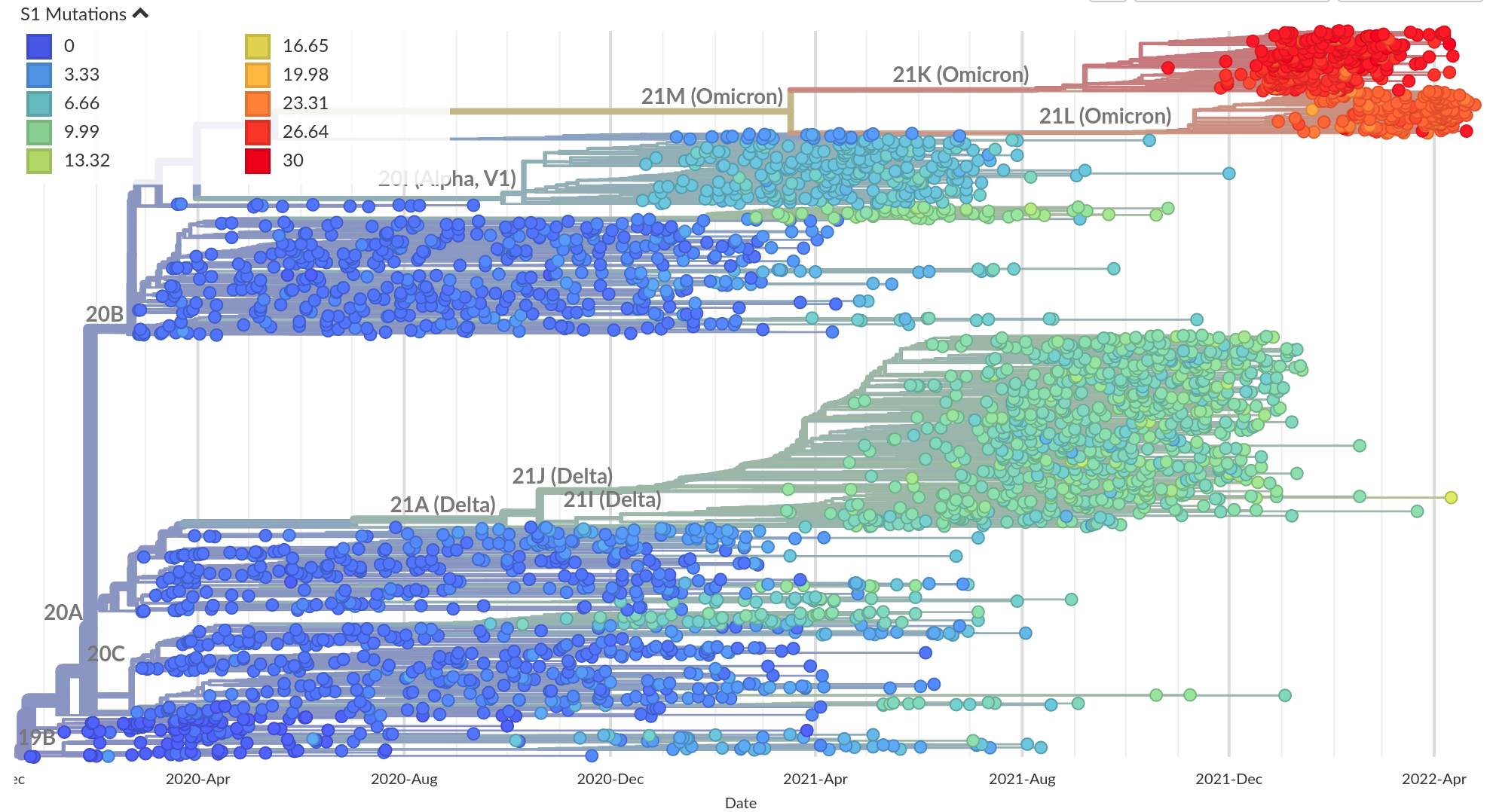
Remarkable patterns of rapid adaptation
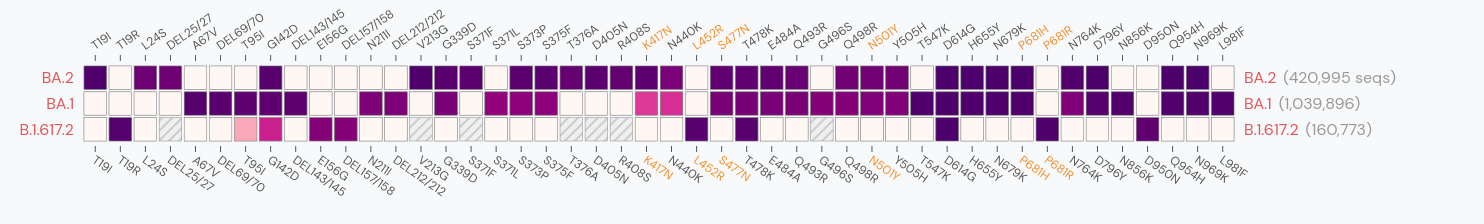
Gradual shift from selection on transmission to immune escape
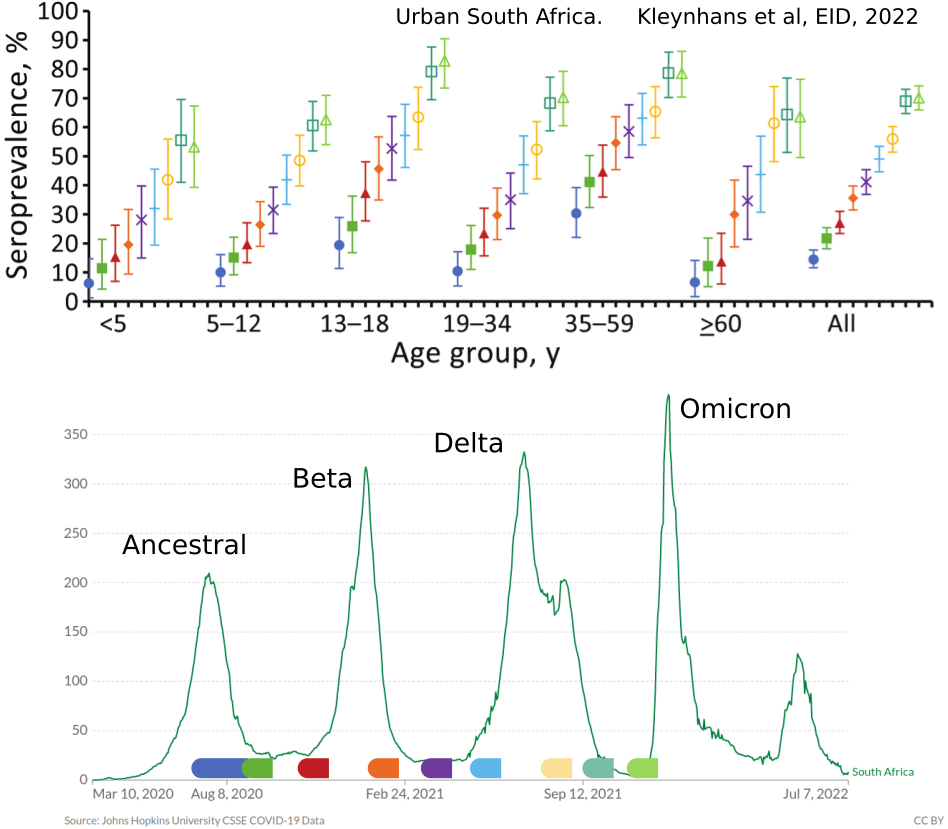
- Until early-2021, seroprevalence was low to moderate
- Delta infections and vaccination resulted in high seroprevalence in 2021
- Variant success now is dominated by immune escape
So far, independent variants have dominated sequentially
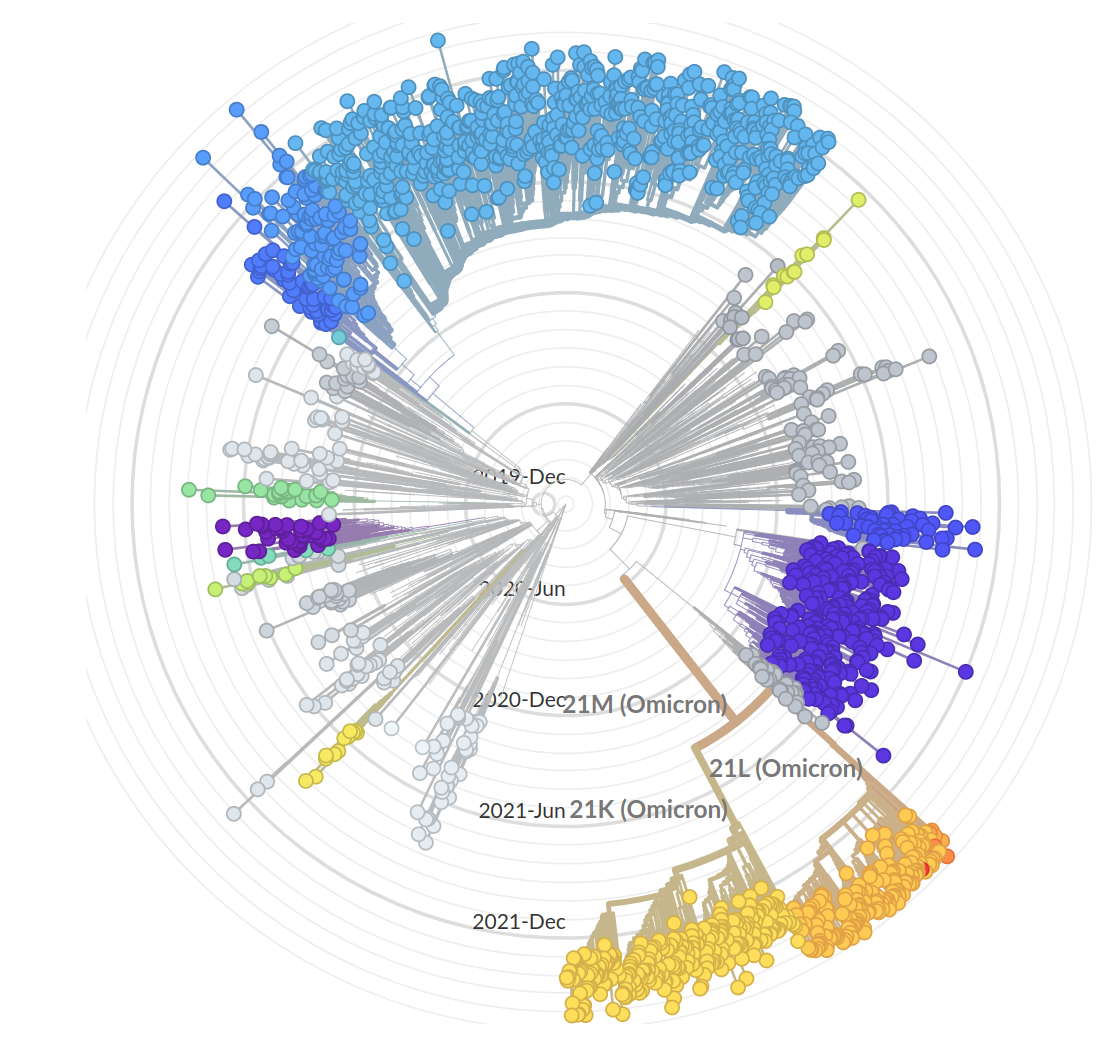
- Variant emergence likely through chronic infections
- Strong dichotomy (until 2022): dramatic changes between variants, slow and steady within variants
- Omicron variants are more dynamic
- With BA.4/5 and BA.2 subvariants, we start to see second generation variants
What is driving this?
Can we predict?
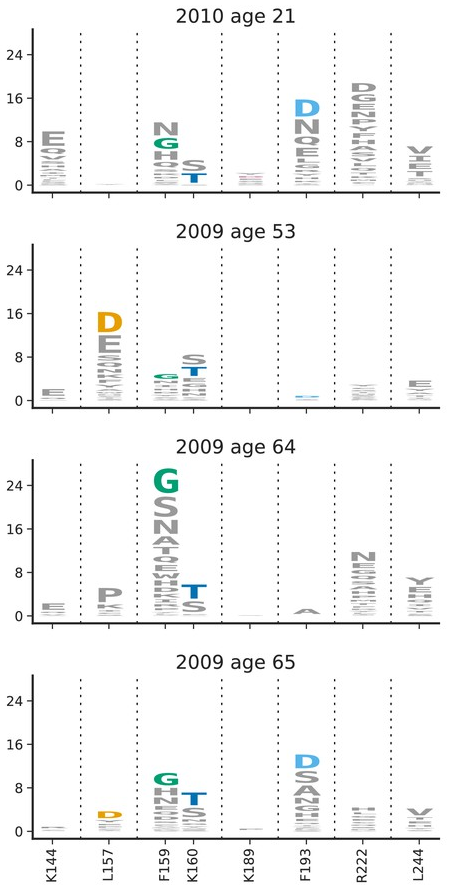
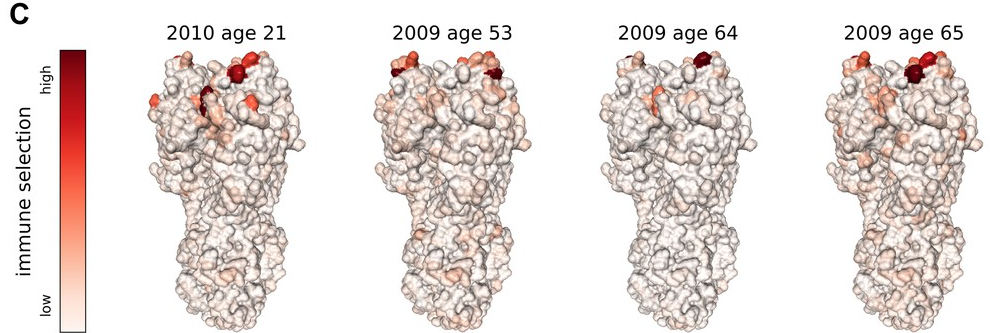
To predict, we need to quantify selection by immunity
- Given a population immunity "landscape", how much escape is caused by which mutation?
- How variable are individual immune responses?
- How does exposure history shape neutralization of different variants?
- What is the contribution of chronic vs acute infections?
- Does escape during chronic infection mediate inter-individual escape? It did for Omicron, but will this stay that way?
- What is the contribution of chronic infections in other viruses?
Summary
- RNA virus evolution can be observed directly
- Rapidly adapting population require new population genetic models
- Those model can be used to infer fit clades
- Future influenza population can be anticipated
- Automated real-time analysis can help fight the spread of disease
HIV acknowledgments







- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
SARS-CoV-2 acknowledgements

.jpg)
- Emma Hodcroft (now in Bern)
- Moira Zuber (Basel)
- Iñaki Comas and Fernando Gonzalez-Candelas, Valencia
- Martina Reichmuth and Christian Althaus (Bern)
- Tanja Stadler, Sarah Nadeau, Tim Vaughan at ETH
- Alberto Hernando and David Matteo at Kido Dynamics
- Jesse Bloom, Katherine Crawford at Fred Hutch
- David Veesler, Alex Walls, Davide Corti, John Bowen at UW
Acknowledgments
Trevor Bedford and his lab -- terrific collaboration since 2014
