Virus evolution and the spread of infectious disease
Richard Neher
Biozentrum, University of Basel
slides at neherlab.org/202311_Ringvorlesung.html
Viruses
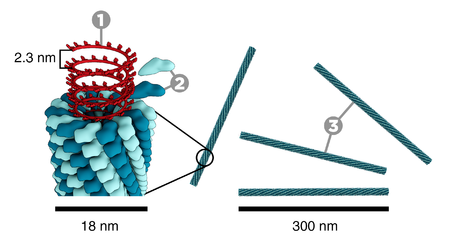 tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
tobacco mosaic virus
(Thomas Splettstoesser, wikipedia)
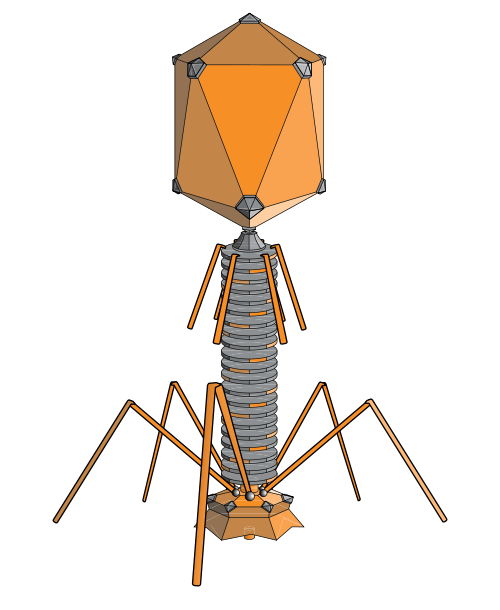
bacteria phage (adenosine, wikipedia)
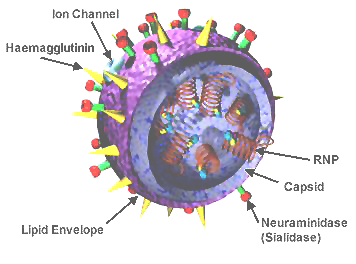
influenza virus wikipedia

human immunodeficiency virus wikipedia
- rely on host to replicate
- little more than genome + capsid
- genomes typically 5-200k bases (+exceptions)
- most abundant organisms on earth $\sim 10^{31}$
Lifecycle of animal viruses
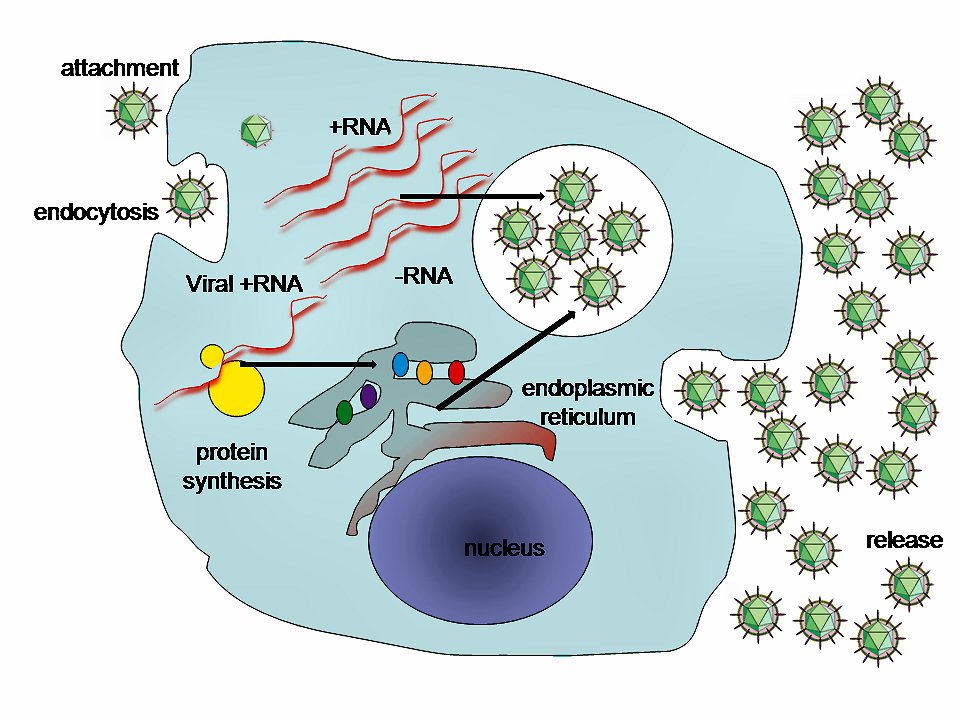 By GrahamColm at English Wikipedia
By GrahamColm at English Wikipedia
Development of sequencing technologies
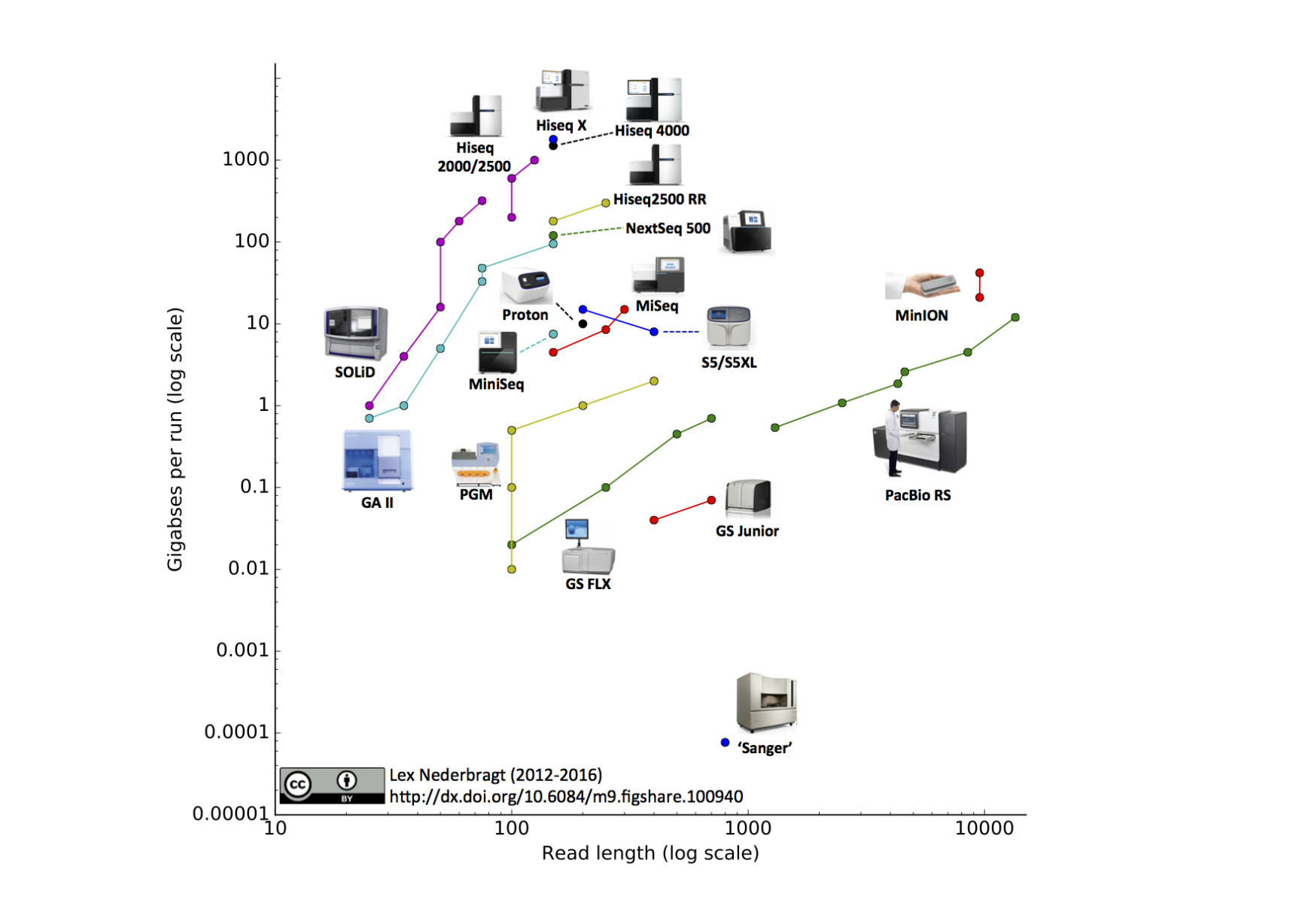
We can now sequence...
- thousands of bacterial isolates
- thousands of single cells
- populations of viruses, bacteria or flies
- diverse ecosystems
Phylogenetic analysis of viral sequences


RNA viruses have a high mutation rate. New mutations arise every few weeks.
Some viruses evolve a million times faster than animals
Animal haemoglobin

HIV protein

Evolution of HIV
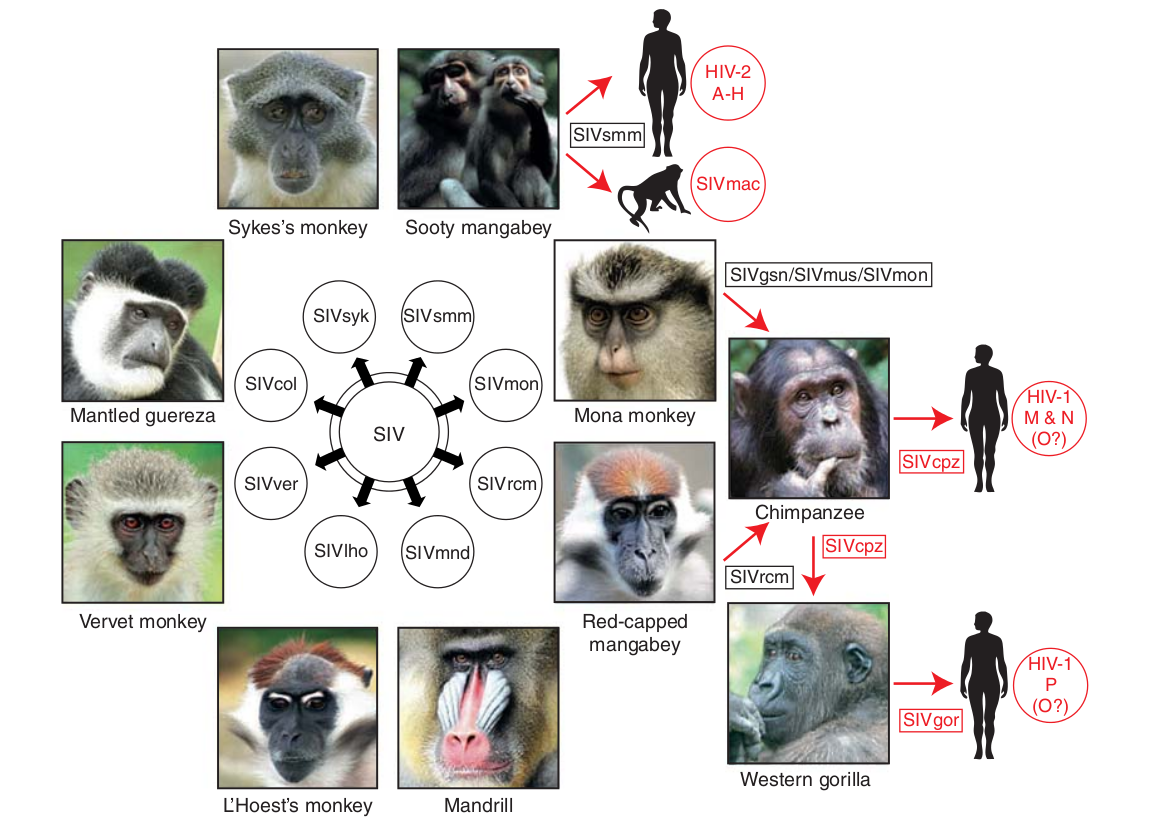
- Chimp → human transmission around 1900 gave rise to HIV-1 group M
- ~100 million infected people since
- subtypes differ at 10-20% of their genome
- HIV-1 evolves ~0.1% per year
HIV infection
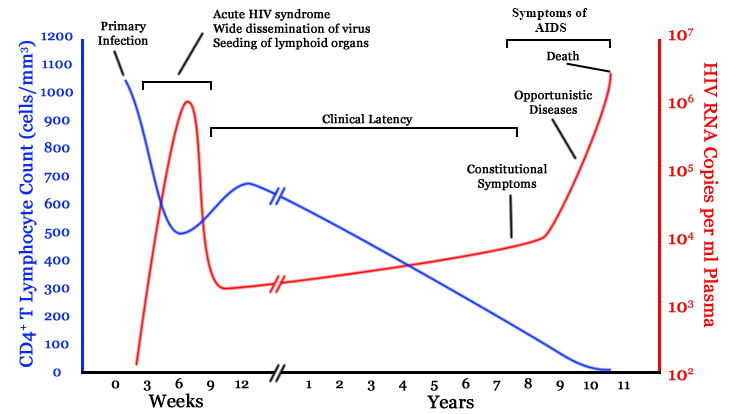

- $10^8$ cells are infected every day
- the virus repeatedly escapes immune recognition
- integrates into T-cells as
latent provirus
{kind=link}
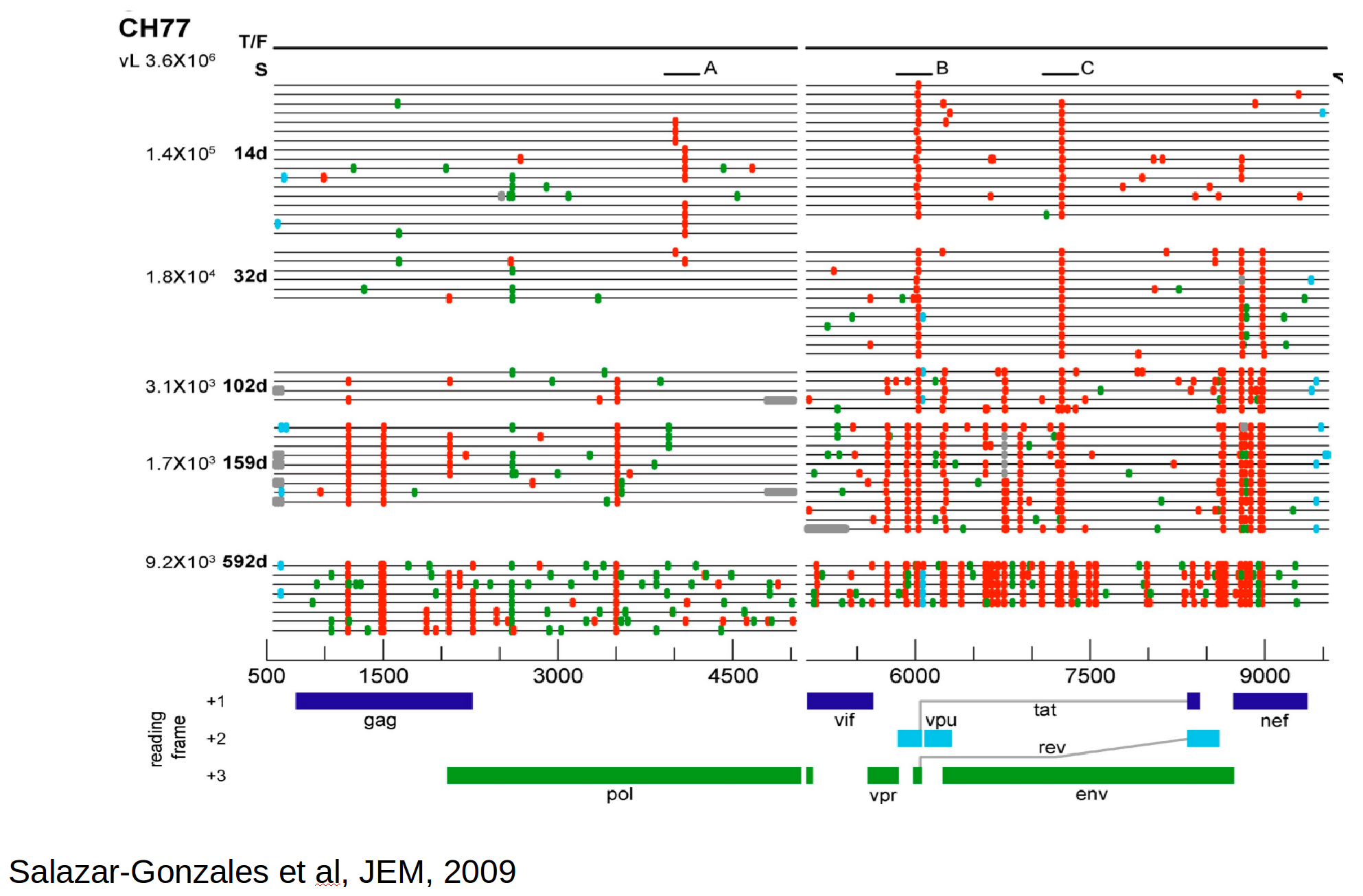
HIV-1 evolution within one individual

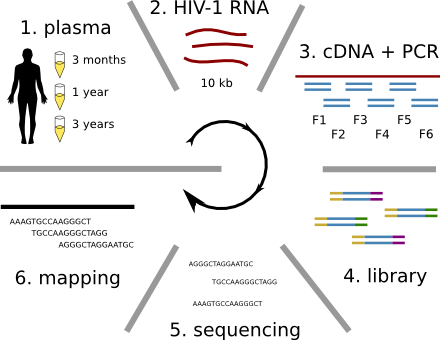

Immune escape in early HIV infection
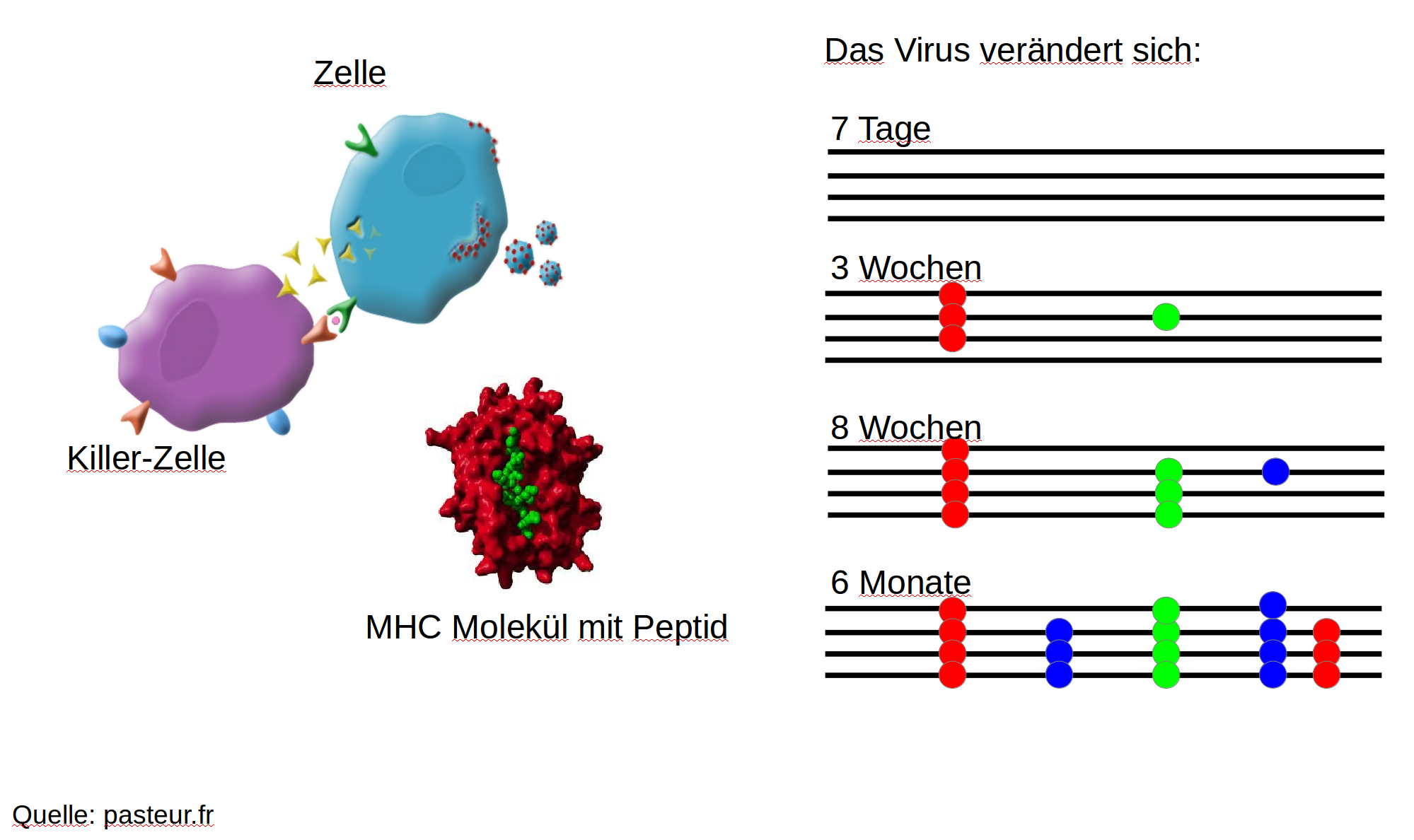
Immune escape in early HIV infection
Selective sweeps
- Viruses carrying a beneficial mutation have more offspring: on average $1+s$ instead of $1$
- $s$ is called selection coefficient
- Fraction $x$ of viruses carrying the mutation changes as $$x(t+1) = \frac{(1+s)x(t)}{(1+s)x(t) + (1-x(t))}$$
- In continuous time → logistic differential equation: $$\frac{dx}{dt} = sx(1-x) \Rightarrow x(t) = \frac{e^{s(t-t_0)}}{1+ e^{s(t-t_0)}}$$
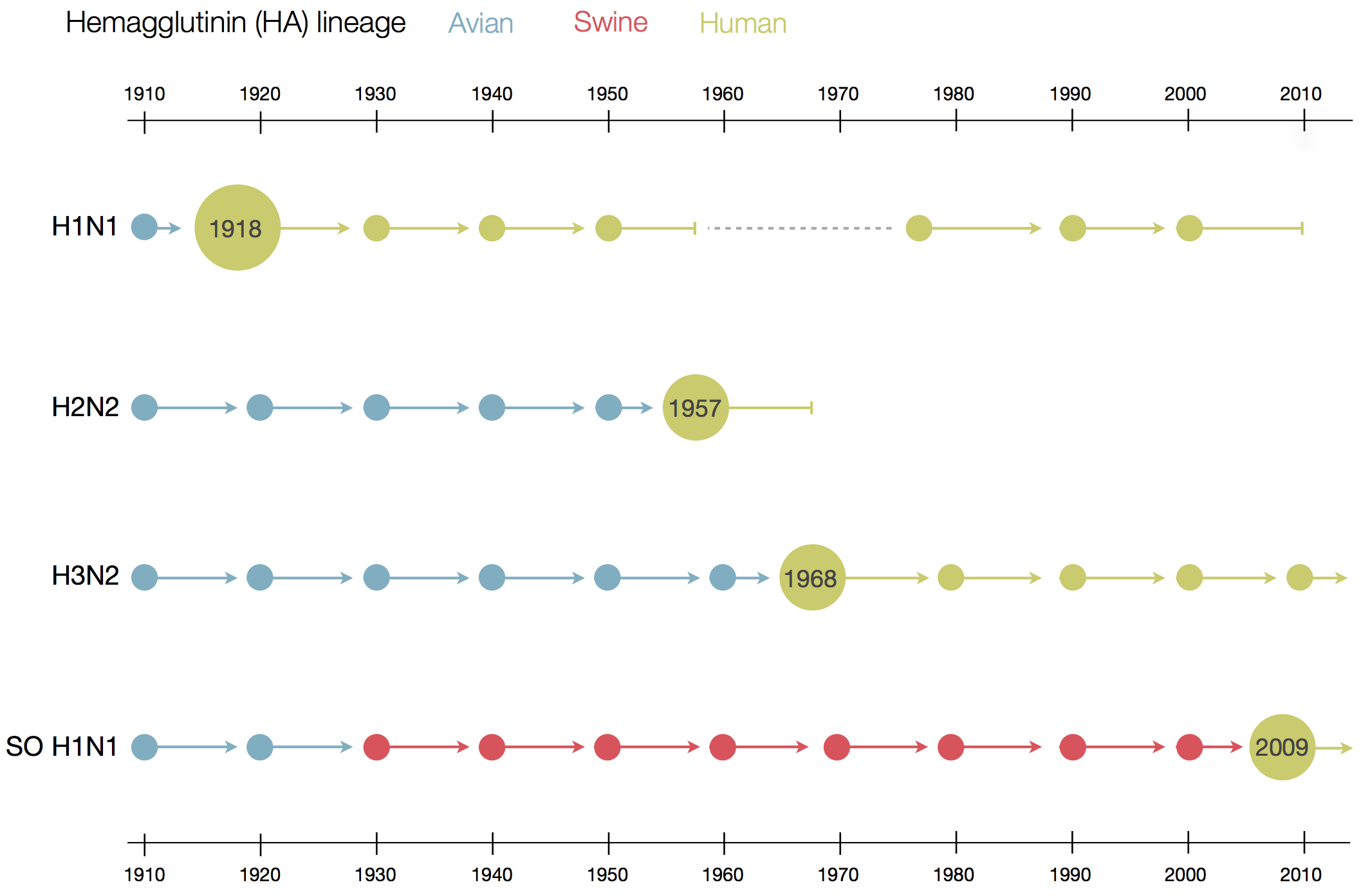
Human Influenza A viruses
Weekly numbers of positive influenza tests in the US by subtype
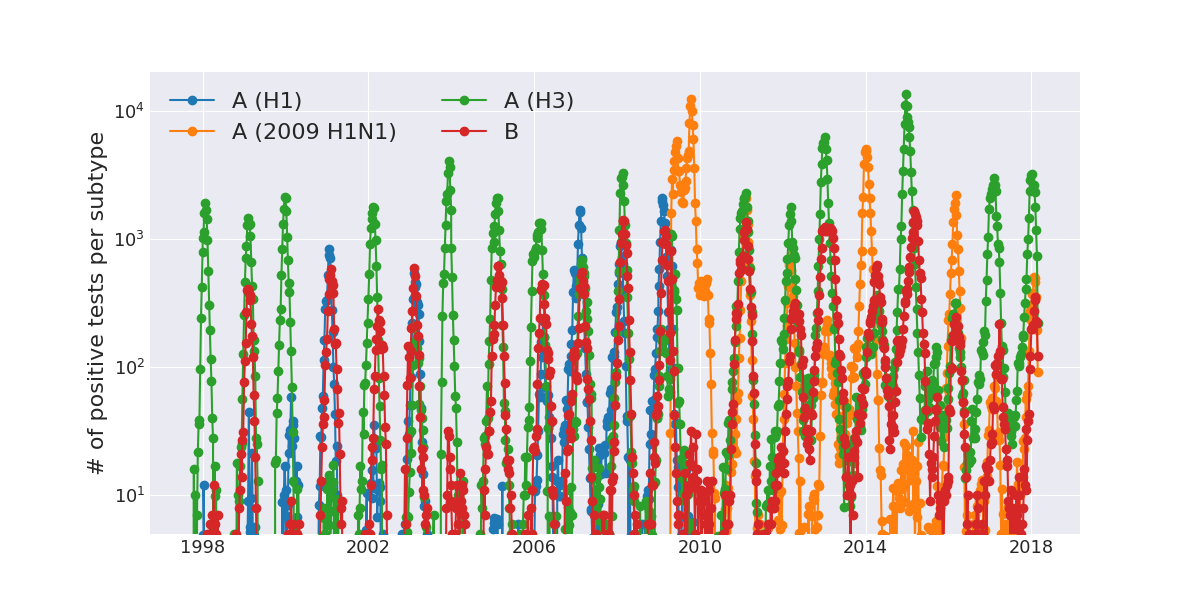 Data by the US CDC
Data by the US CDC
Influenza virus
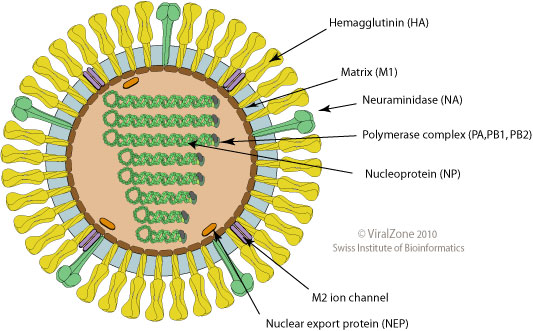
- Surface proteins hemagglutinin (HA) and neuraminidase (NA)
- Influenza A virus
- Common in birds and mammals
- Many different subtypes defined by surface proteins
- H3N2, H1N1, H7N9, H5N1
- Influenza B virus
- infects mainly humans
- two lineages that split 30-40y ago
- B/Victoria vs B/Yamagata
nextflu.org
joint work with Trevor Bedford & his lab
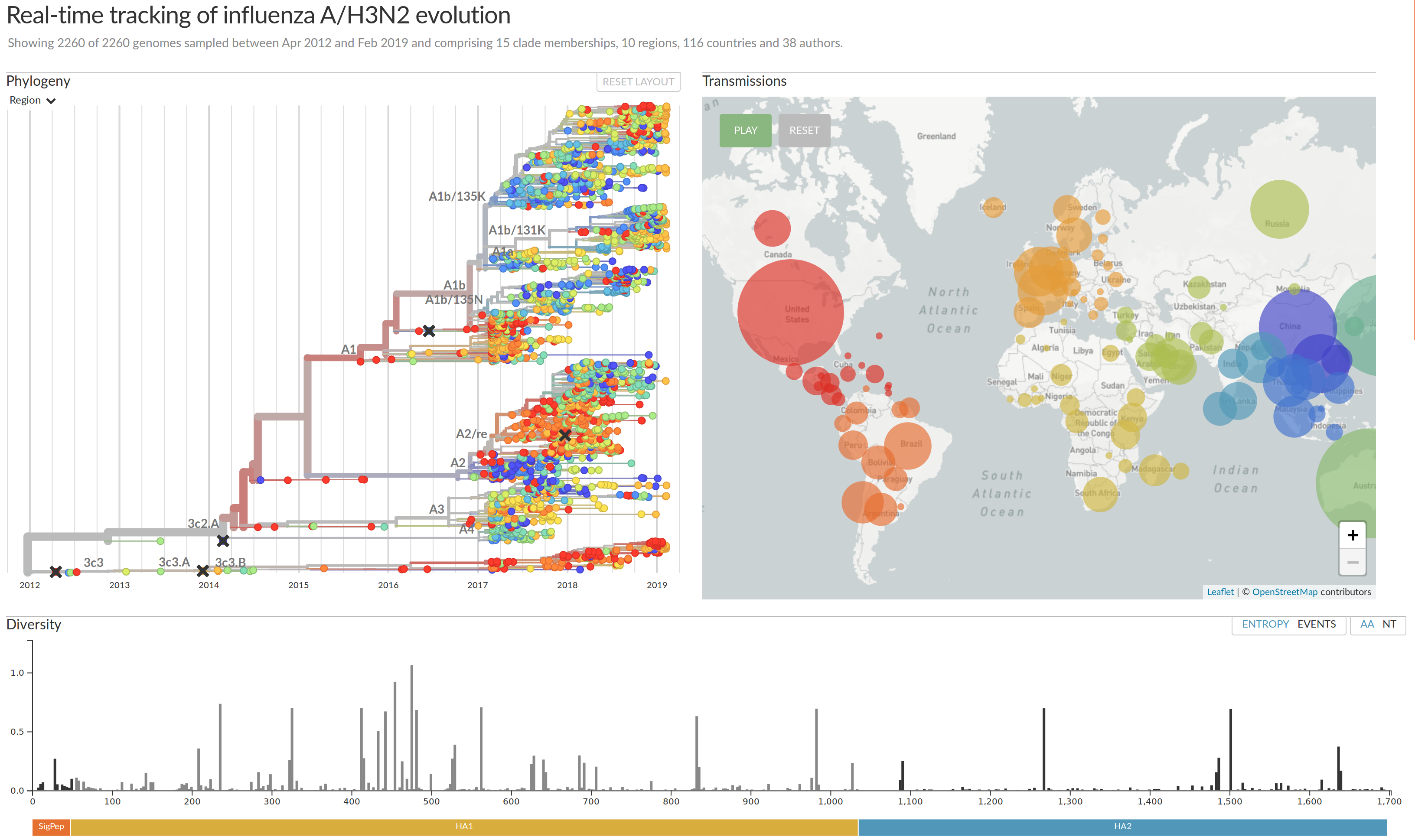
nextstrain.org
joint work with Trevor Bedford & his lab
Clonal interference and traveling waves
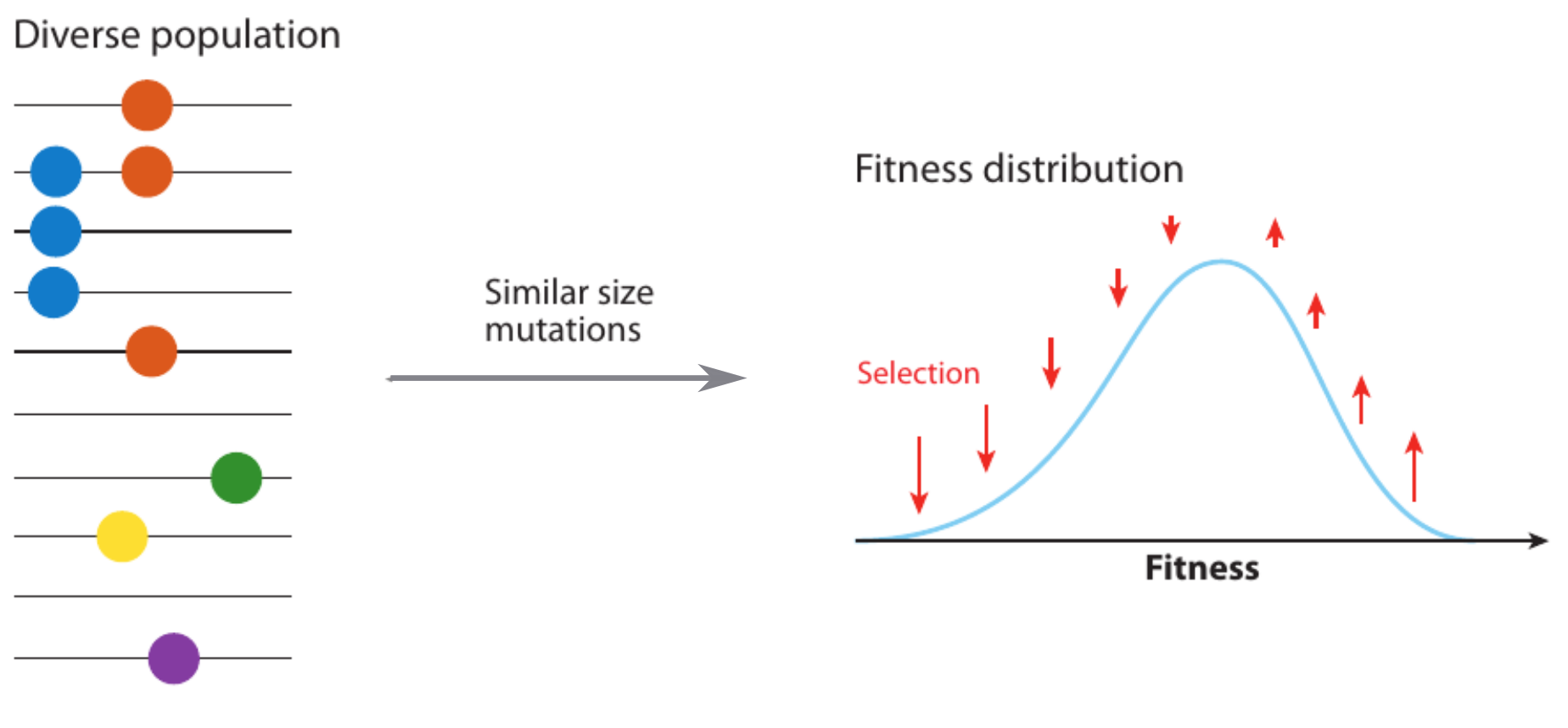
Typical tree
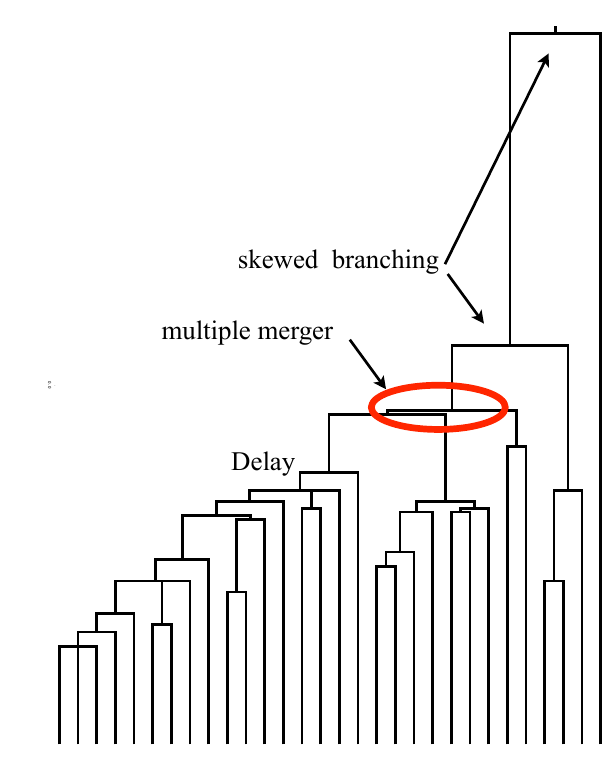
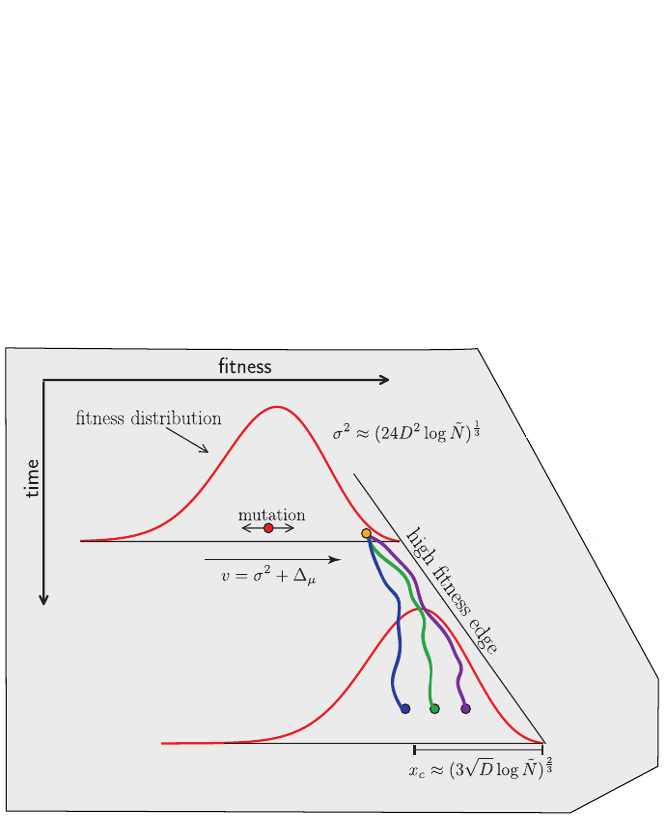
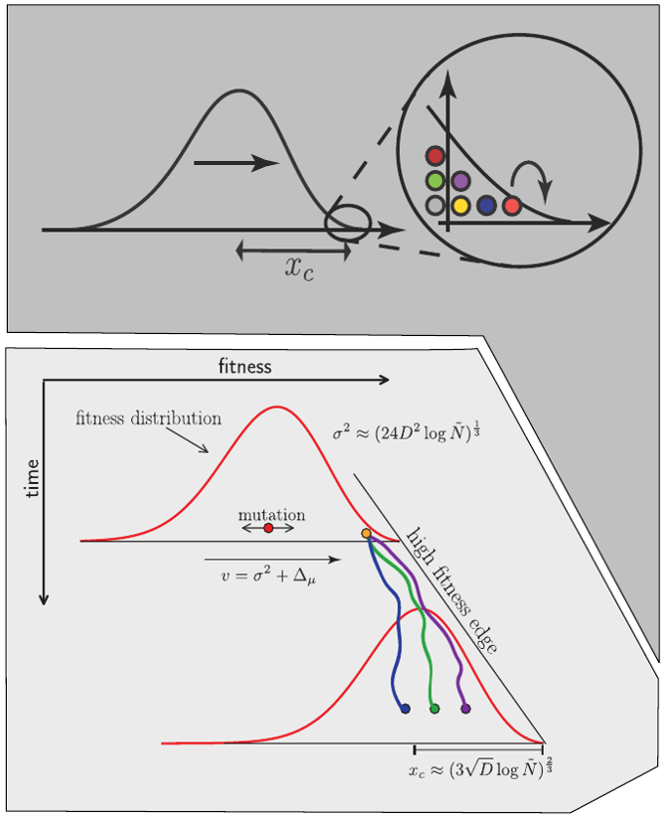
Bolthausen-Sznitman Coalescent
Bursts in a tree ↔ high fitness genotypes
Can we read fitness of a tree?
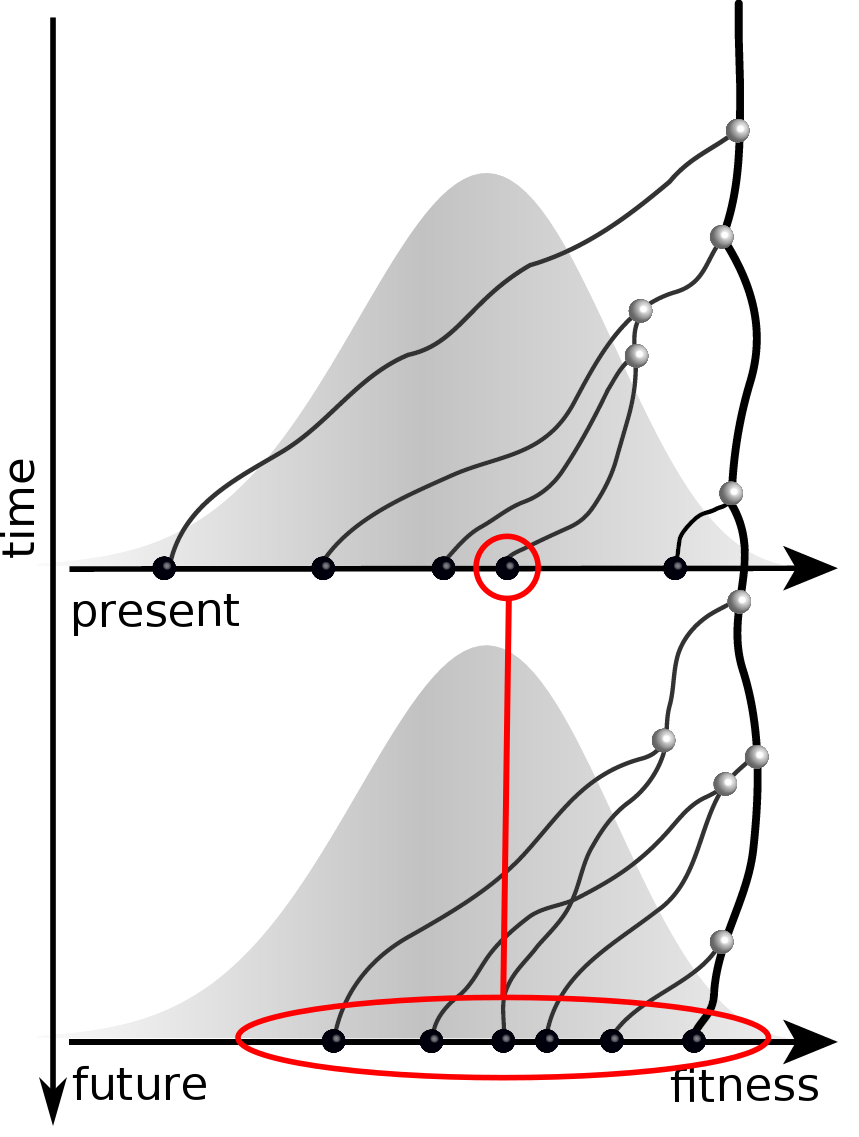
Predicting evolution
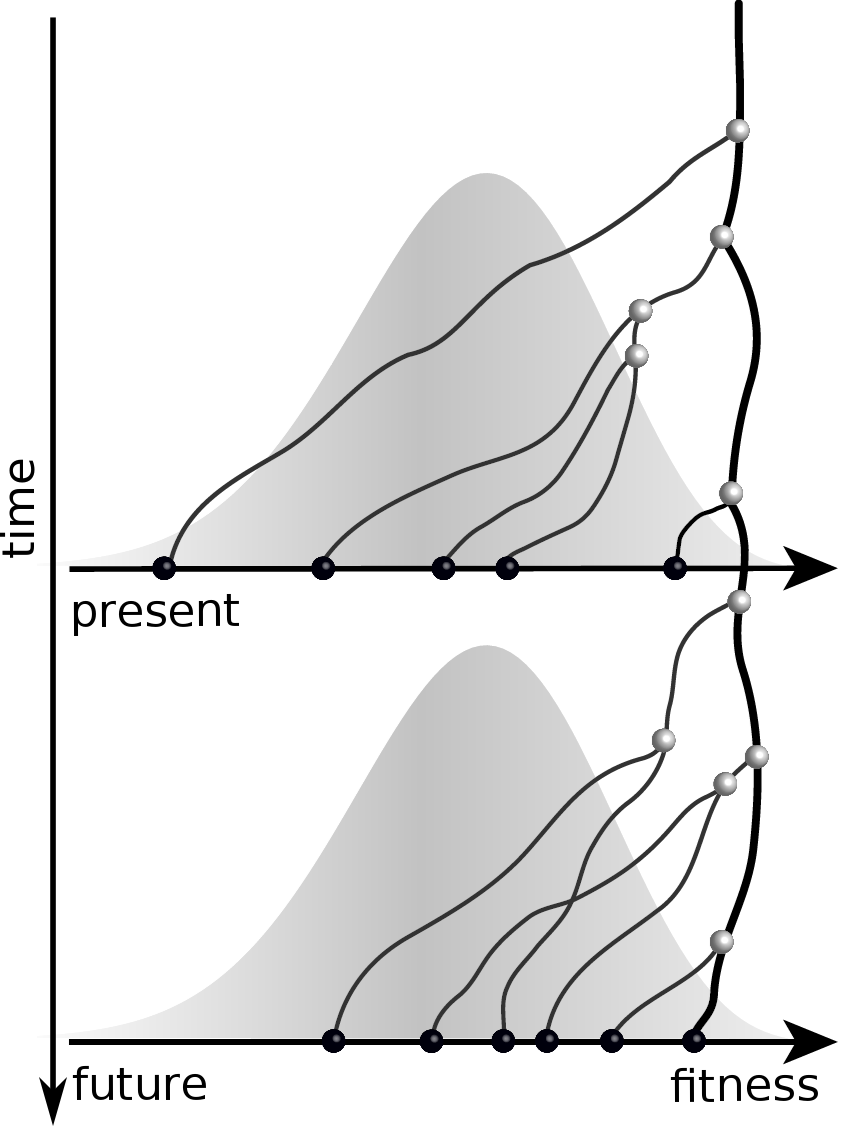
Given the branching pattern:
- can we predict fitness?
- pick the closest relative of the future?
Validation on simulated data
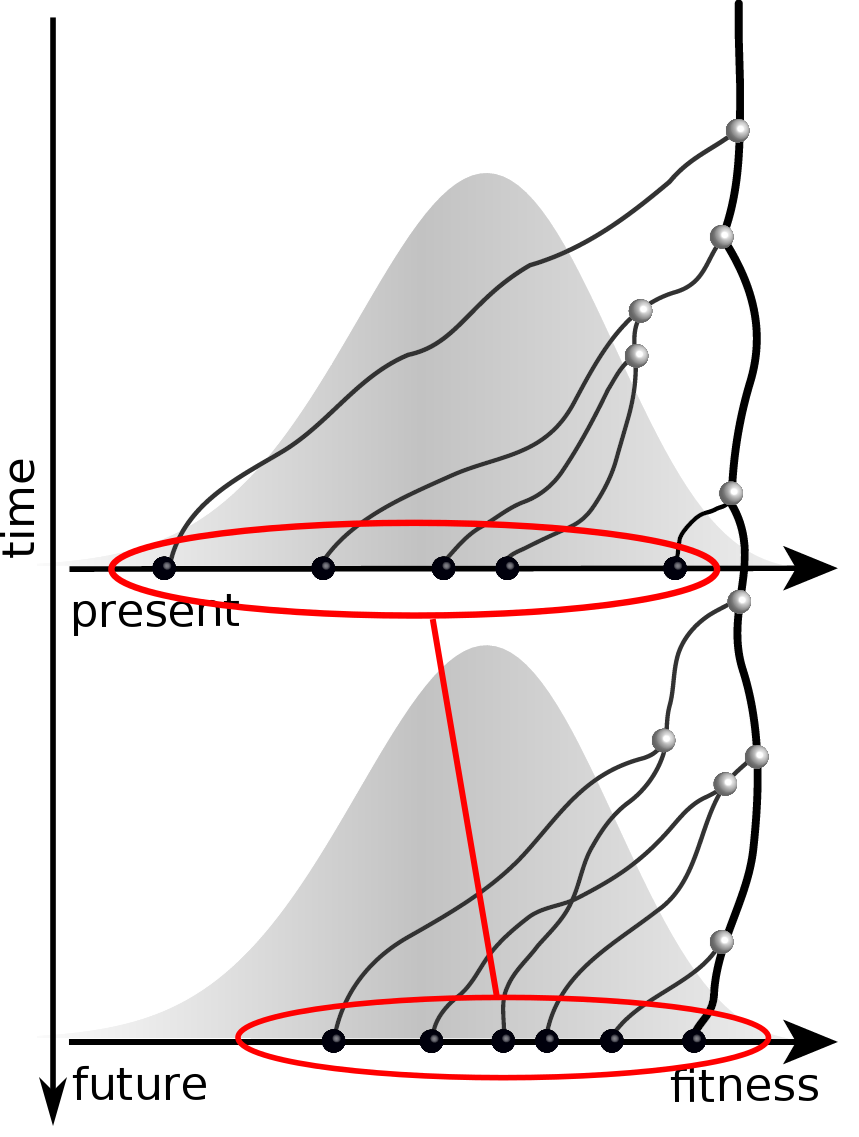
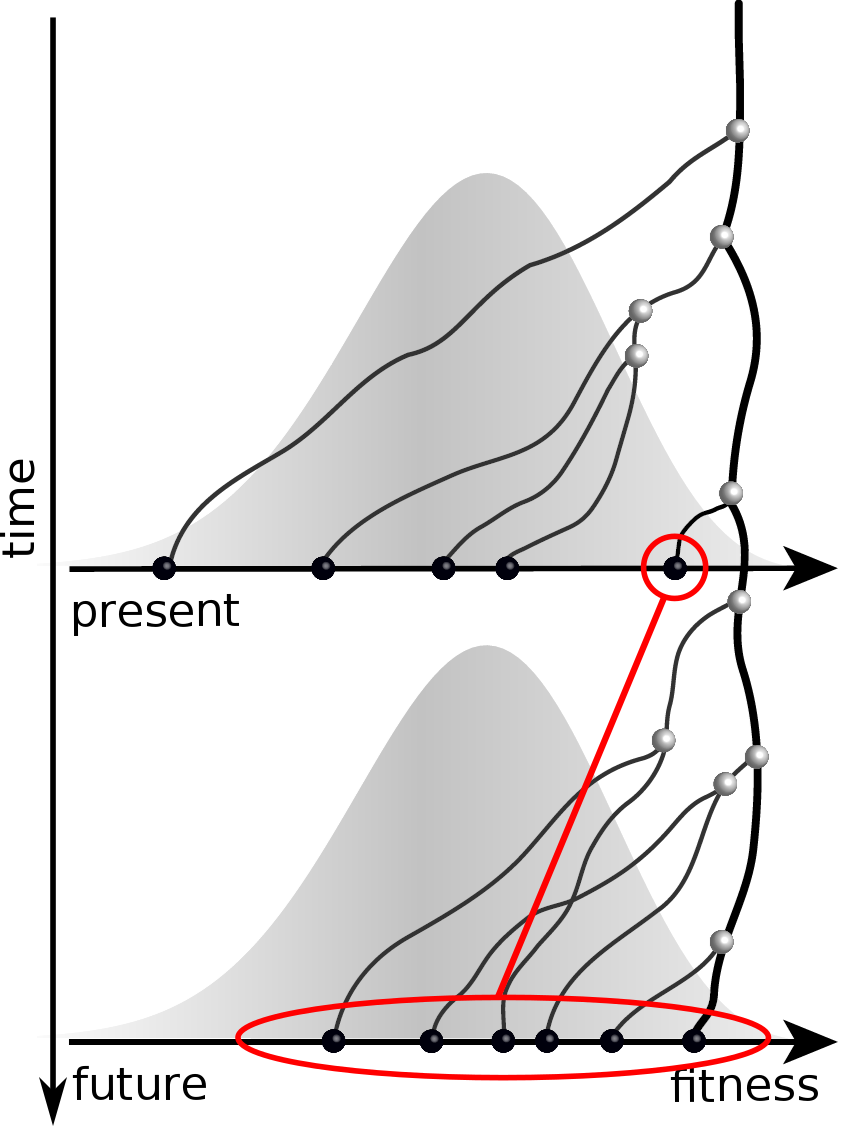
Prediction of the dominating H3N2 influenza strain
- no influenza specific input
- how can the model be improved? (see model by Luksza & Laessig)
- what other context might this apply?
Summary
- RNA virus evolution can be observed directly
- Rapidly adapting population require new population genetic models
- Those model can be used to infer fit clades
- Future influenza population can be anticipated
- Automated real-time analysis can help fight the spread of disease
HIV acknowledgments







- Fabio Zanini
- Jan Albert
- Johanna Brodin
- Christa Lanz
- Göran Bratt
- Lina Thebo
- Vadim Puller
Influenza and Theory acknowledgments




- Boris Shraiman
- Colin Russell
- Trevor Bedford
- Oskar Hallatschek
Acknowledgments
Trevor Bedford and his lab -- terrific collaboration since 2014
