Update: the paper is now published.
A few days ago, I uploaded a revision of our recent manuscript (with Taylor Kessinger and Boris Shraiman) on genetic diversity in sexual populations under selection. I would like to elaborate a little bit on what I think is remarkable about our results.
Why is it important? It is common these days to sequence multiple individuals from a population and analyze the genetic diversity in the sample to learn something about demographic and evolutionary past. To infer the past from diversity data, we need to know how diversity depends on the parameters and processes we are interested in. This link typically comes from the analysis of simple models. The predominant framework used for this purpose is the neutral coalescent, which is often used as a null model to detect selection. This strategy -- looking for outliers in a mostly neutral genome -- seemed like a good strategy at the time when it was thought that the great majority of polymorphims are neutral. If, however, the majority of polymorphisms is under some form of selection, we need a new null model to detect adaptations of particular interest that stand out from all the rest that, while not neutral, has weak or fluctuating effects. Our manuscript aims at delivering such a null. In contrast to previous analysis that focussed on mutations with strong effects (background selection or hitch-hiking), we analyze a model where a large number of weakly selected polymorphisms generate fitness diversity in a sexual population. We find that the properties of neutral diversity smoothly interpolate between the neutral limit (drift dominated) and the limit of strong selection (draft dominated). The crossover between the two regimes happens when fitness difference between haplotypes are comparable to the inverse population size. The length of haplotypes (LD) and the diversity are self-consistently determined and depend on the fitness variance per maplength, but only weakly on the population size. To determine where a population sits on this continuum between neutral or draft dominated regime, it is informative to analyze the site frequency spectrum, which changes qualitatively between the regimes.
How did we address it?
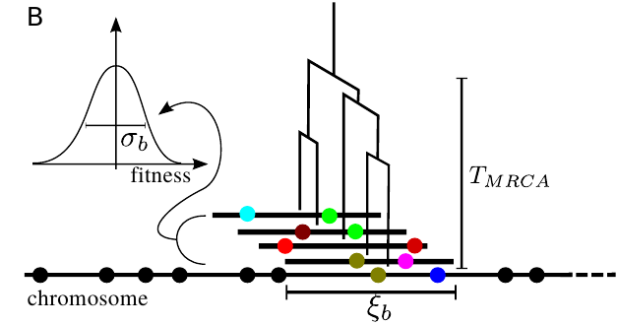
In sexual populations, crossing over reshuffles alleles, which results
in linkage equilibrium and independent histories of loci at large
distances. The histories of tightly linked loci, however, remain
correlated and very close loci behave as if they were asexual. These
different degrees of linkage interact with selection in complicated
ways. Our approach to this problem was to identify the length of blocks
that behave more or less asexually over the time to the most recent
common ancestor at the locus, calculate the fitness variation within
those blocks that, and map the problem to results for coalescence with
selection in asexual populations.
 {
.wp-image .alignleft width="370"
height="195"}
{
.wp-image .alignleft width="370"
height="195"}
The latter problem has been addressed by Oskar Hallatschek and myself. We showed that in asexual populations with substantial selected diversity, coalescence and genetic diversity are not described by the Kingman (standard neutral) coalescent, but resemble the Bolthausen-Sznitman coalscent (BSC) -- at least in the limit of large populations. Michael Desai, Aleksandra Walczak and Daniel Fisher published similar conclusions.
What's next? It is common to define an "effective population size", Ne, via the distance between pairs of haplotypes and hope that a neutral model with this Ne explains other features of genetic diversity. This rarely works. Furthermore, Ne depends strongly on crossover rates, functional density (purifying selection), etc. The one quantity Ne is only weakly correlated with is the census population size. Our results link genetic diversity (I refuse to call it Ne) to parameters such as mutation rates, crossover rates, and effect distributions of mutations. The predictions should be applicable whenever there are many polymorphisms within a linkage block, which is likely the case in facultative outcrossers or low recombination regions of obligate outcrossers.
When analyzing resequencing data, it should be possible to use the polarized site frequency spectrum to determine whether diversity is dominated by drift or draft. In the draft regime, heterozygosity should be proportional to the square root of of rho/mu s\^2, where rho is the crossover rates, mu is the mutation rate, and s\^2 is the average squared effect of mutations.