We have a new preprint on the arXiv
(here on Haldane's
sieve).
This work is the result of a collaboration between us and Alan
Perelson, LANL, and explores methods to
estimate parameters of the HIV-immune system interaction from time
resolved sequence data. The focus of this paper is on early
infection
 {.alignright width="281"
height="204"}
dominated by a few rapid substitutions that fix because they prevent or
reduce recognition of infected cells by the immune system via cytotoxic
T-lymphocytes (CTL). CTL escape is one of the fastest instances of
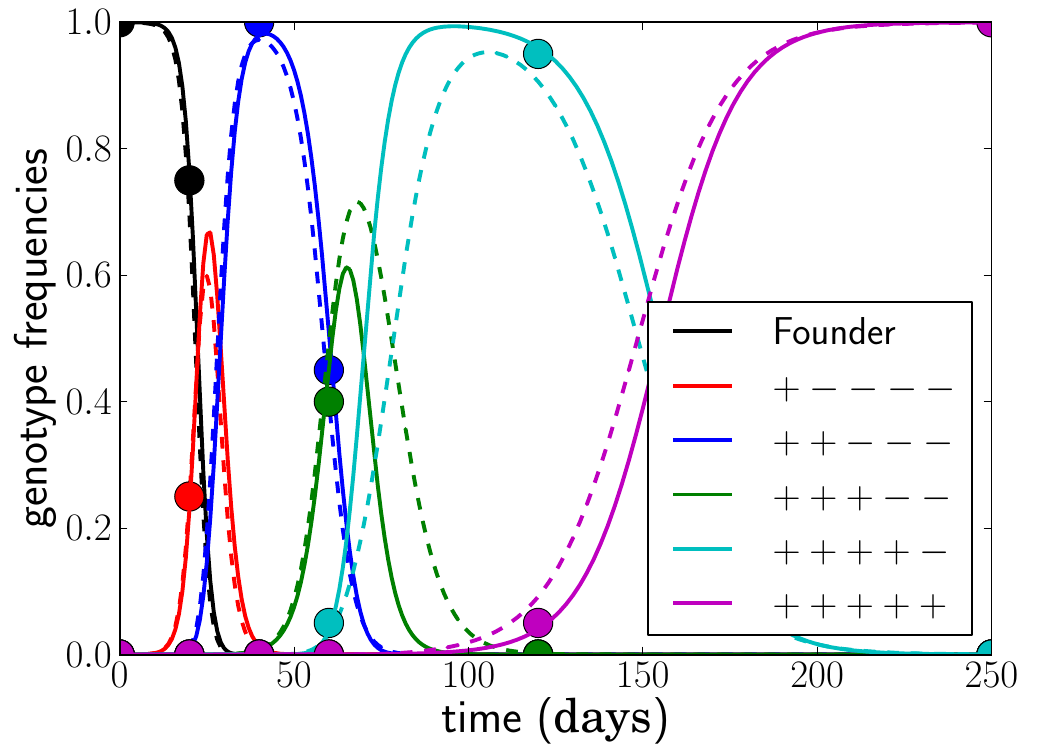
evolution I have come across. 4-6 mutations spread within a few weeks.
It happens in most HIV infections and is partly predictable based on the
HLA genotype of the infected person. These substitutions are so rapid
that clonal interference has to be modeled. Our method fits a reduced
model of clonal interference to the typically very sparse data and
thereby estimates the selection coefficients, aka escape rates.
{.alignright width="281"
height="204"}
dominated by a few rapid substitutions that fix because they prevent or
reduce recognition of infected cells by the immune system via cytotoxic
T-lymphocytes (CTL). CTL escape is one of the fastest instances of
evolution I have come across. 4-6 mutations spread within a few weeks.
It happens in most HIV infections and is partly predictable based on the
HLA genotype of the infected person. These substitutions are so rapid
that clonal interference has to be modeled. Our method fits a reduced
model of clonal interference to the typically very sparse data and
thereby estimates the selection coefficients, aka escape rates.
Why do we want to know these numbers? The number of viruses in the blood of an infected person peaks 2-3 weeks after infection and thereafter drops by 2-3 order of magnitude. This drop is partly due to a response by the adaptive immune system. However, it has proved difficult to attribute this drop to specific parts of the immune response. The rates at which different mutations sweep through the population gives us information about the pressure exerted by the T-cell clones that target the epitope containing this mutation.
How do we do it? Early in infection, the viral population is large and selection is strong. In these conditions, recombination is of minor importance since most double/triple... mutants are more efficiently produced by recurrent mutation than recombination. This implies that mutations accumulate sequentially always on a background one which already all previous mutations are present. The time at which a novel mutation happens in tightly constrained by the trajectory of preceding genotype. These constraints regularize the fitting problem to some degree and the multi-locus fitting is more robust than single locus fitting.
What do we learn about evolution in general? In addition to the intrinsic interest in the HIV/CTL interaction, CTL escape is an ideal setting to study rapidly evolving populations. This evolution happens in its "natural" habitat and the selective pressure as well as the functional consequences of the observed molecular changes can be quantified via immunological data, protein structure, and replication assays. In addition, we have ample cross-sectional data (HIV sequences from many different patients) that allows us to look at prevalence of the escape mutations and potential compensatory mutations. None of this is done in this paper, but studying HIV/immune-system coevolution is a fascinating show case of rapid evolution.