Fabio and Richard just published a paper in Journal of Virology about whether or not synonymous mutations in HIV are neutral. We know of course that many synonymous sites in HIV have important roles in regulation and RNA structure, but what about those sites at which we see high-frequency synonymous polymorphisms without known function? We focus on the env gene and in particular on the region containing the V loops that are under antibody attack by the immune system and hence change rapidly (V stands for variable). We investigate the dynamics of synonymous mutations in serial sequence data and show that most synonymous mutations in that region come with a fitness cost of the order of 0.002 / day. Nevertheless, they seem to hitch-hike to high frequency on linked adaptive variants. These synonymous mutations interfere with folding of the HIV RNA molecule, reducing its replication efficiency.
Why is this important?
In studies of molecular evolution, synonymous mutations are often used as neutral control used to infer selection operating on the proteins. Our results add to the growing body of evidence that selection on synonymous sites needs to be accounted for when doing such analysis. This not only holds for viruses, but has recently been demonstrated in the fruit fly.
More importantly, we shed light on the functional relevance and evolutionary dynamics of RNA structures in the genome. A recent study compared RNA structures of diverged strains of HIV and found that the overall pattern of folding is conserved, while the exact base pairs are not. Our results can explain this observation: most RNA structures seem to be important to the virus, but not crucial (disrupting mutations have a small fitness effect). This results in occasional fixation of a disrupting mutation which is slowly restored by compensatory mutations or rearrangements. This way the molecular architecture of the RNA structure can change leaving the overall pattern in place.
How did we do it?
In longitudinal HIV sequence datasets, we observe new synonymous alleles
rising in frequency in our sample up to 50% and higher (see sketch), but
almost all of them disappear from our sample after one to two years
(i.e., the ancestral allele is found again in all sequences). This
suggests that these mutations are deleterious. We quantify this effect
by measuring how long it takes for these mutations to disappear and how
often they do not disappear.
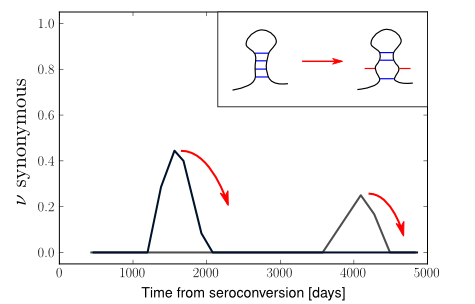
Comparing these observations to extensive computer simulations, we conclude that the majority of synonymous mutations are deleterious with small fitness effects (0.002 / day). Then, building on published RNA pairing data, we ask whether or not a mutation that destroys a base pair is pruned more often than any other mutation; that seems to be the case indeed.