My review on "Genetic draft, selective interference, and population genetics of rapid adaptation" in Annual Reviews of Ecology, Evolution, and Systematics is finally out (not exactly final yet, some notational issues will be corrected). Sally Otto had asked me to write an accessible summary of the work published over the last 10-15 years on adaptation and selective interference. Some of this work was done by scientists with backgrounds in physics like myself. Owing to differences in notation and mathematical approaches, population geneticists sometimes struggled with these papers. Coming from physics and having worked in population genetics for 6 years, I have tried to synthesize this work in a streamlined and accessible fashion -- let me know if it worked. To illustrate some of the ideas, I have put together a website with some python scripts that simulate different scenarios discussed in the paper: http://webdav.tuebingen.mpg.de/interference/
Drift vs Draft
Classical population genetics emphazises the competition between
stochastic effects in reproduction (genetic drift) and deterministic
forces such as selection. In idealized models, genetic drift stems from
non-heritable randomness in offspring number. The width of this
offspring number distribution is assumed (very) small compared to the
population size and the law of large numbers garantees that many similar
models converge to the same diffusion limit where the strength of drift
is inversely proportional to the population size. However, a different
source of randomness is often much more important: random associations
to genetic backgrounds of different fitness result in background
selection, Hill-Robertson effects, and selective interference.
 {.wp-image-172
.alignright width="400"
height="166"}
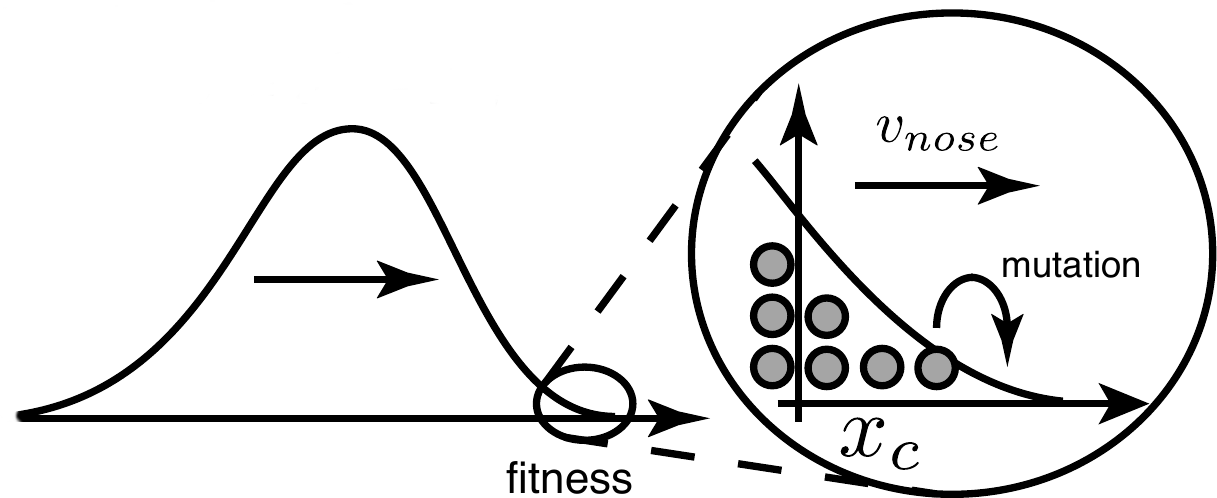
While the effect of background fitness on allele frequencies might be weak in
a single generation, associations to genetic backgrounds are (partly)
heritable and the effects amplify over many generations. This
amplification is multiplicative and the resulting differences in
offspring number after several generations can be comparable to the
population size. In other words, the effective offspring distributions
after several generations are very skewed with long power-law tails. In
fact, these distributions can be so broad that the variance diverges
with the population size. In this case, no diffusion limit is possible
and the statistical properties of drift and and linked selection are
fundamentally different.
{.wp-image-172
.alignright width="400"
height="166"}
While the effect of background fitness on allele frequencies might be weak in
a single generation, associations to genetic backgrounds are (partly)
heritable and the effects amplify over many generations. This
amplification is multiplicative and the resulting differences in
offspring number after several generations can be comparable to the
population size. In other words, the effective offspring distributions
after several generations are very skewed with long power-law tails. In
fact, these distributions can be so broad that the variance diverges
with the population size. In this case, no diffusion limit is possible
and the statistical properties of drift and and linked selection are
fundamentally different.
Asexual vs sexual The effects of draft are strongest in asexual organisms where the entire chromosome stays linked forever. However, linked selection can also be substantial in facultatively species such as plants, worms, yeasts or viruses (think influenza). As soon as there is the potential for the rapid expansion of a particular line (be it because of an intrinsic fitness advantage or favorable environmental conditions in a particular spot), the effective "many-generation" offspring distribution can become very broad and draft dominates over drift. In obligatly sexual species, the effects of draft are confined to the chromosomal neighborhood, but linkage to alleles at different distances still gives rise to stochastic forces very different from the classical genetic drift (rare tight linkage to a beneficial allele essentially sweeps one haplotype to fixation, loosely linked sweeps only bounce it around a little).
Recent Developments: Genealogical methods for rapid adaptation Many successful population genetic methods have used the duality between Kimura's diffusion models and the Kingman coalescent. This duality allows the efficient computation of statistics by considering the backward process of observed alleles, rather than the forward process of the entire population. Recent developments suggest that a similar duality exists for models dominated by draft: Genealogies in these models share statistical properties with a particular coalescent process known as Bolthausen-Sznitman coalescent that allows for multiple mergers. This coalescent process can predict a number of observable features in sequence data such as the site frequency spectra, the time to the most recent common ancestor, etc. I briefly discuss these very recent results in the review.
Why should we care? You might say "Let's just define an effective population size and pretend all linked selection is some sort of drift". But many population genetic methods detect outliers above a random background. To detect outliers reliably, we need to understand the null distribution. The background has very different statistical properties when the dominant source of randomness is draft rather than drift and using the wrong null model will reduce the power of the test and produce false positives. In other applications, one estimates values of parameters of simple models and these models better capture the relevant population genetic processes. It is for example popular to estimate the history of the effective population size from the rate of coalescence in the past. In many cases, in particular for large populations under selection, this effective population size has very little to do with the actual population size. Instead, one estimates the rate of coalescence which depends on the relative success of different lineages, which in turn depends on fitness, environmental fluctuations, and luck.