In this preprint -- a collaboration with Colin Russell and Boris Shraiman -- we show that it is possible to predict which individual from a population is most closely related to future populations. To this end, we have developed a method that uses the branching pattern of genealogical trees to estimate which part of the tree contains the "fittest" sequences, where fit means rapidly multiplying. Those that multiply rapidly, are most likely to take over the population. We demonstrate the power of our method by predicting the evolution of seasonal influenza viruses.
How does it work? Individuals adapt to a changing environment by accumulating beneficial mutations, while avoiding deleterious mutations. We model this process assuming that there are many such mutations which change fitness in small increments. Using this model, we calculate the probability that an individual that lived in the past at time t leaves n descendants in the present. This distributions depends critically on the fitness of the ancestral individual. We then extend this calculation to the probability of observing a certain branch in a genealogical tree reconstructed from a sample of sequences. A branch in a tree connects an individual A that lived at time \(t_A\) and had fitness \(x_A\) and with an individual B that lived at a later time \(t_B\) with fitness \(x_B\) as illustrated in the figure. B has descendants in the sample, otherwise the branch would not be part of the tree. Furthermore, all sampled descendants of A are also descendants of B, otherwise the connection between A and B would have branched between \(t_A\) and \(t_B\). We call the mathematical object describing fitness evolution between A and B "branch propagator" and denote it by \(g(x_B, ,t_B |x_A,t_A)\).
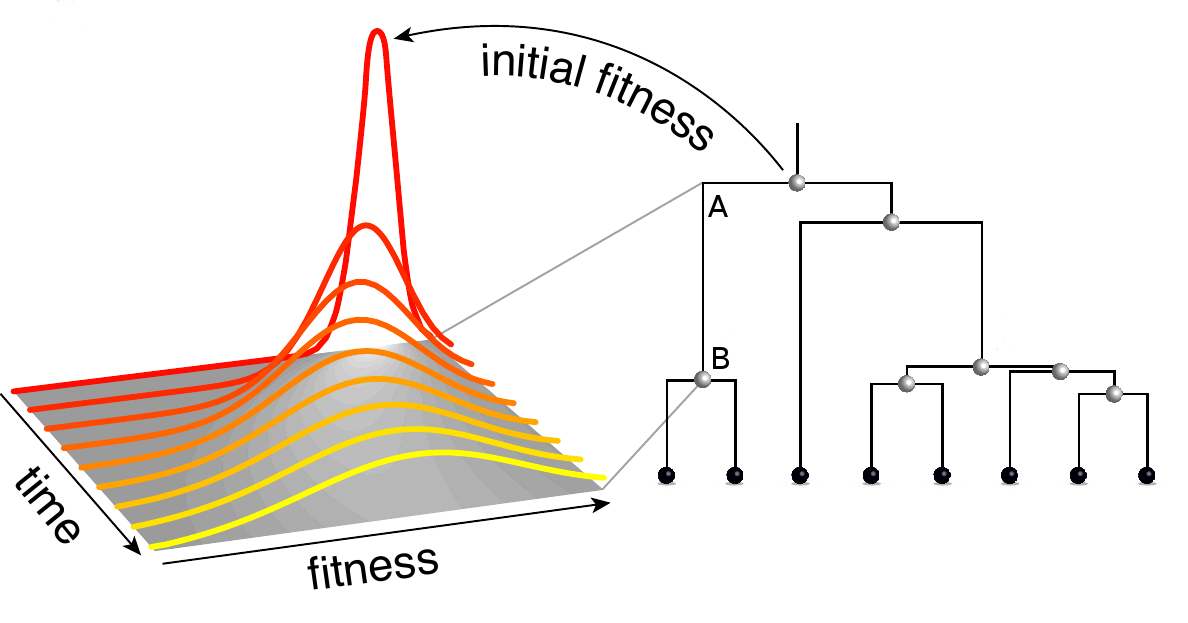
The joint probability distribution of fitness values of all nodes of the tree is given by a product of branch propagators. We then calculate the expected fitness of each node and use it to rank the sampled sequences. The top ranked sequence is our prediction for the sequence of the progenitor of the future population.
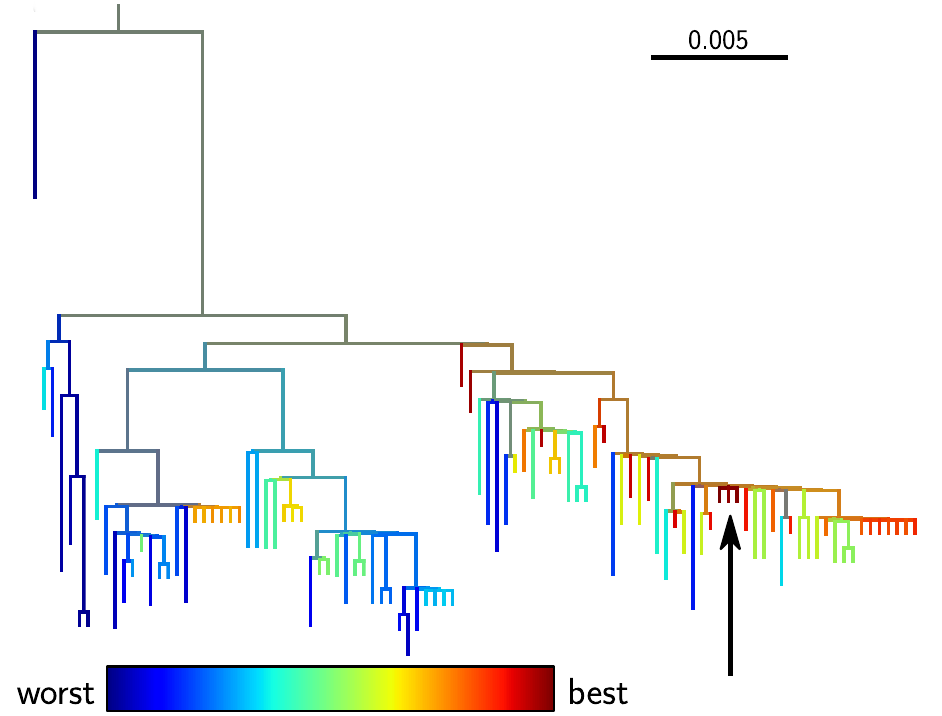
Why do we care? Being able to predict evolution could have immediate applications. The best example is the seasonal influenza vaccine, that needs to be updated frequently to keep up with the evolving virus. Vaccine strains are chosen among sampled virus strains, and the more closely this strain matches the future influenza virus population, the better the vaccine is going to be. Hence by predicting a likely progenitor of the future, our method could help to improve influenza vaccines. One of our predictions is shown in the figure, with the top ranked sequence marked by a black arrow.
Influenza is not the only possible application. Since the algorithm only requires a reconstructed tree as input, it can be applied to other rapidly evolving pathogens or cancer cell populations. In addition, to being useful, the ability to predict also implies that the model captures an essential aspect of evolutionary dynamics: influenza evolution is to a substantial degree -- enough to enable prediction -- dependent on the accumulation of small effect mutations.
Comparison to other approaches Given the importance of good influenza vaccines, there has been a number of previous efforts to anticipate influenza virus evolution, typically based on using patterns of molecular evolution from historical data. Along these lines, Luksza and Lässig have recently presented an explicit fitness model for influenza virus evolution that rewards mutations at positions known to convey antigenic novelty and penalizes likely deleterious mutations (+a few other things). By using molecular influenza specific signatures, this model is complementary to ours that uses only the tree reconstructed from nucleotide sequences. Interestingly, the two models do more or less equally well and combining different methods of prediction should result in more reliable results.