In our new preprint, we report whole genome deep sequencing of longitudinally sampled HIV-1 populations from multiple patients -- effectively a movie of evolution at about 6 month resolution. This work was led by Fabio and is the product of a fantastic collaboration with the group ofJan Albert at the Karolinska Institute in Stockholm.
Among the many things we can study in detail using this data set, we looked at linkage and recombination. We find that linkage disequilibrium in chronic infection is typically limited to about 100bps. Consistent with this lack of long range linkage, the shapes of trees reconstructed from 400bp reads varies greatly in different regions of the genome. 400bp are often too short to construct well supported phylogenetic trees. Nevertheless, the trees are instructive to illustrate diversity in the population. The figure below animates trees when moving through genome from 5' to 3' end.
Every position in genome has a unique genealogical tree, but through recombination genealogical trees of two sites diverge as the distance between the sites increases. One way to picture this process is to think of a book in which each page show the genealogy corresponding to a particular nucleotide. Skimming through the book results in a movie of gradually changing trees. We need diversity to resolve trees and can't reconstruct a tree for an individual site, but the trees obtained from sliding 400bp windows approximate this process.
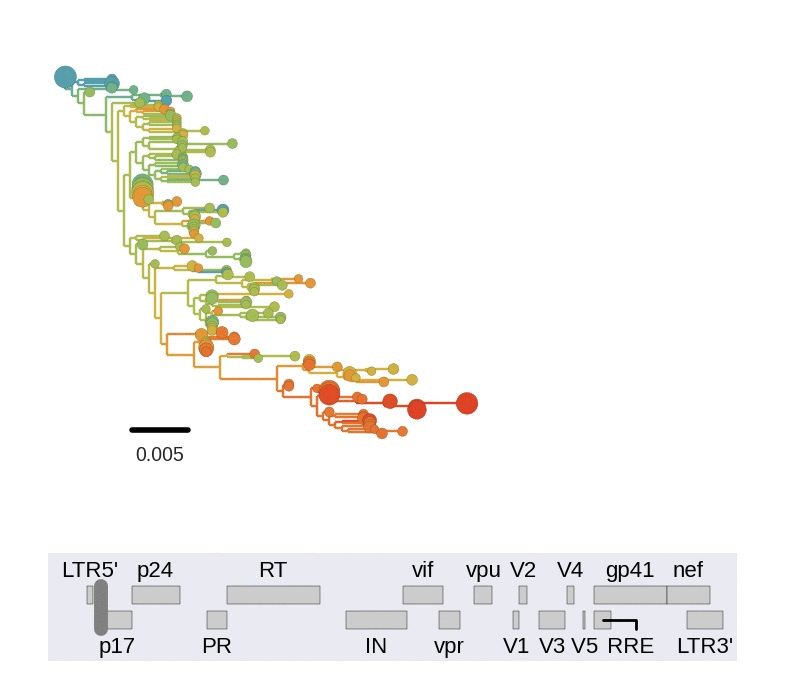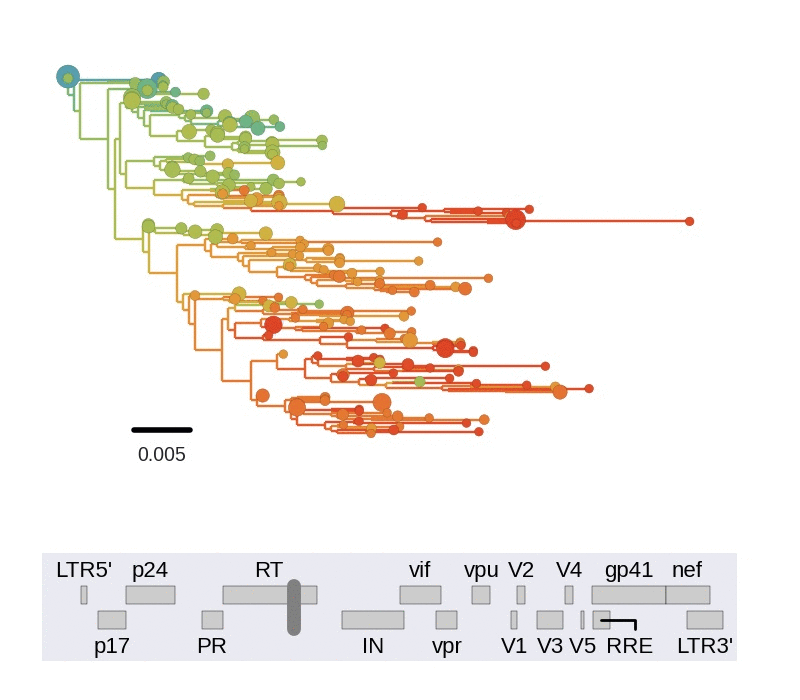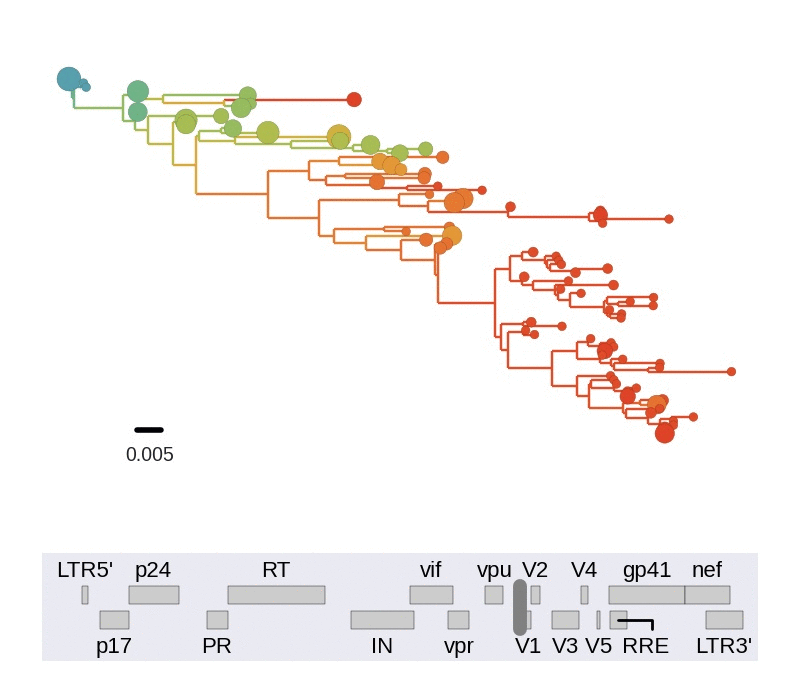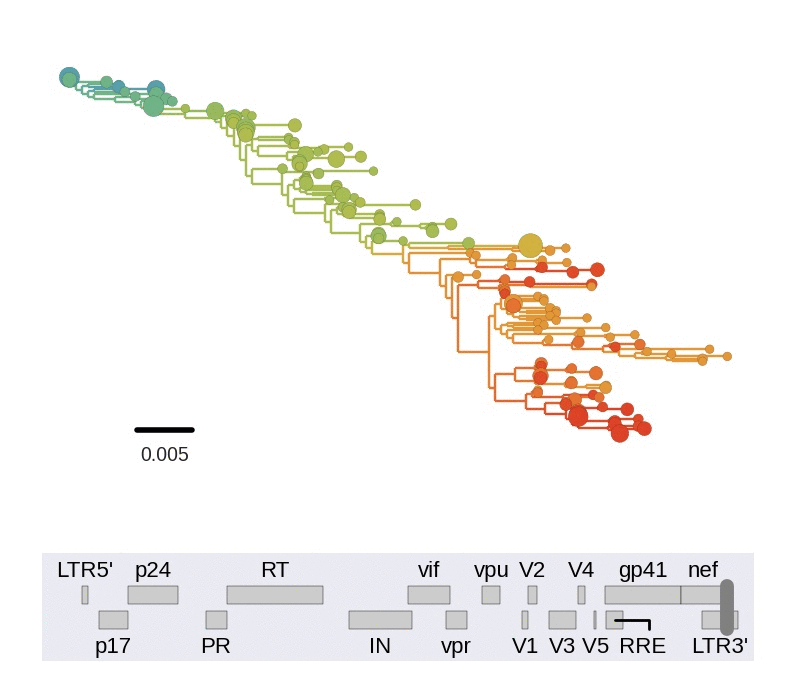 Trees of longitudinally sampled sequences in various parts of the HIV-1
genome. Big circles correspond to common variants, small circles to rare
variants. Early samples are shown in blue, followed by green, yellow and
red.[/caption]
Trees of longitudinally sampled sequences in various parts of the HIV-1
genome. Big circles correspond to common variants, small circles to rare
variants. Early samples are shown in blue, followed by green, yellow and
red.[/caption]
Trees in different parts of the genome vary widely in shape and depth. This is consistent with extensive recombination. The scale of linkage -- about 100bp -- is compatible with earlier estimates of the intrapatient recombination rate by us and Thomas Leitner or Batorsky and colleagues.