Within HIV infected individuals, the human immune system tries to prevent virus replication while HIV continuously changes to avoid recognition by the immune system. The resulting evolution of the virus population has become a paradigmatic example of rapidly adapting populations. We just published a paper that provides unprecendented insight into intrapatient HIV evolution. This work is the product of an extremely enjoyable collaboration involving Fabio Zanini from our group here in Tuebingen and the group of Jan Albert at the Karolinska Institute in Stockholm.
Whole genome deep sequencing
Our aim in this study was to provide a comprehensive assessment of the evolutionary dynamics unfolding within the body of HIV infected people. We developed a strategy to sequence the entire virus genome such that even rare mutations are accurately represented in our data set. The impressive sample collections in Sweden and the generous participation of patients in this study allowed us to follow HIV evolution densely in multiple patients. We developed an interactive web application that allows users to explore HIV evolutionary dynamics and access the data in a convenient way.
What did we find?
Mutations occur at random and while selection for replication weeds out harmful mutation and amplifies useful ones. The common conception is that mutation rates are low and/or useful mutations are rare. Furthermore, biological reality is complicated and predicting what might be a useful mutations seems hopeless. In HIV, however, we find a high degree of reproducibility and predictability, indicating that "finding the right mutation" is not a rare, fortunate event for HIV but rather a fast and reliable mechanism of survival.
The predictability extends to single positions in the genome. More then 20% of sites that are globally unconserved (such as sites at which mutations are synonymous) are measurably diverse after a few years within each patient. This diversity is growing continuously with little signs of loss of diversity through genetic drift or hitch-hiking with beneficial mutations. This implies a large population that systematically explores sequence space. In contrast, at conserved sites, we observe next to no diversity indicating efficient selection against deleterious variants.
We can not only predict where mutations accumulate because they are
tolerated, but also where they spread because they help the virus. By
looking specifically at sites where the virus population was initially
different from the majority of HIV sequences known, we found
that the virus has a strong tendency to come back to the global consensus
state.
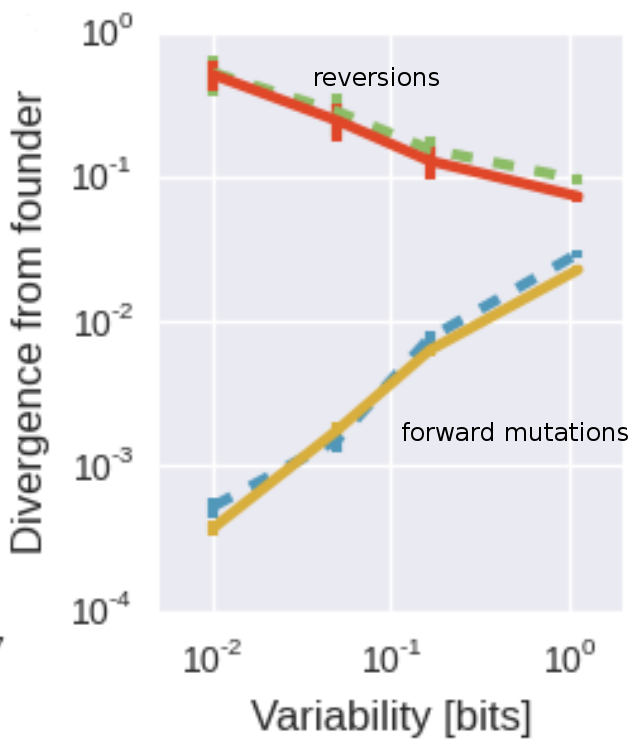
30% of all substitutions occur at the 5% of sites were the initial virus differed from this consensus and represent reversions. The tendency to revert to this global attractor is stronger at sites that are globally more conserved. Within the diversity of HIV-1, this attractor seems universal. The picture on the right shows the rate of evolution (divergence after 6 years) separately sites that can revert and sites already in the consensus state. At the most globally most conserved sites, about 50% of all non-consensus positions revert to consensus after 5 years -- a roughly 1000 fold excess over evolution away from consensus. We also found that reversions are happening not only soon after infection, but rather all along, for many years.
What does this mean?
Our data are consistent with HIV as a large population that systematically explores a mostly universal fitness landscape and returns to favoured state when possible. The reproducible patterns of evolution are only possible since HIV recombines extensively within patients -- without recombination it would be much more difficult for the virus population to simultaneously revert and escape in different regions of the genome as these mutations would interfere with each other as they spread. Sweeping of adaptive mutations would wipe out diversity and the reproducible patterns of mutation accumulation. The reproducibility of minor variation further suggests that the fitness costs of individual mutations are similar among unrelated viruses and explains why inference of fitness landscapes of HIV from cross-sectional data is possible.