The genome of influenza viruses consists of eight segments. Two of these segments, HA and the NA segments, code for proteins on the surface of the virus. These proteins are recognized by the human immune system and change rapidly to evade preexisting immunity. Most phylogenetic and epidemiological analysis is done separately on either the HA or the NA segment. A joint analysis of HA and NA or all eight segments should allow a more accurate reconstruction of influenza spread and in addition will yield information on rates of coinfection or evolutionary interactions between segments.
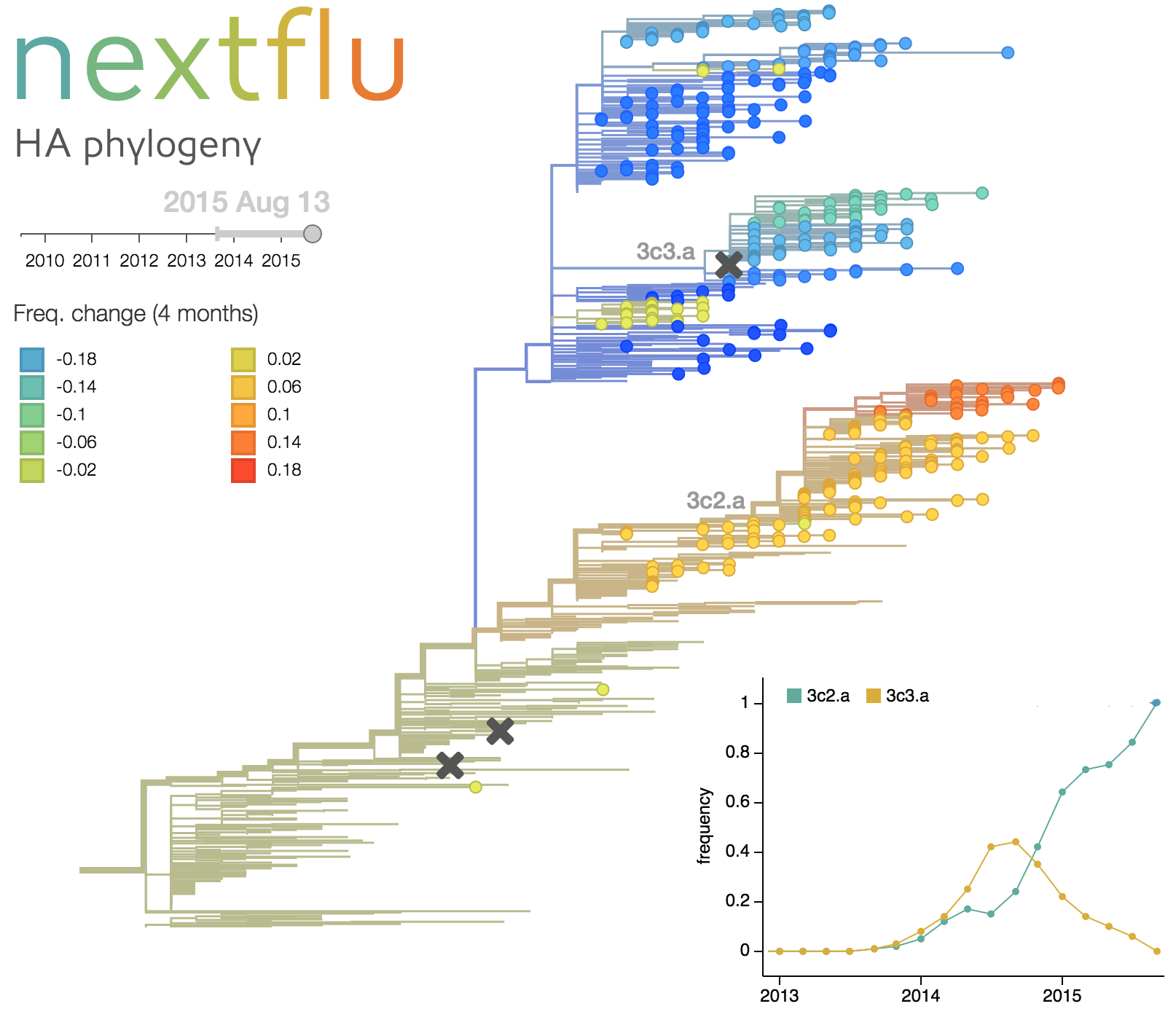
This analysis is to be build on our existing efforts to track, predict, and analyse influenza virus evolution (joint work with the lab of Trevor Bedford at the FHCRR in Seattle). These are implemented as automated pipelines that are rerun automatically as soon as new data are available. The results exposed via interactive web applications at nextstrain.org and nextflu.org.
During the course of this project, the student would become familiar with and/or work on
- probabilistic phylogenetic inference
- population genetics
- coalescent theory
- methods for tree comparison
The project will be directly supervised by Richard Neher. Our interdisciplinary group combines methods and ideas from statistical physics with bioinformatics, population genetics, and phylogenetics. More information about our research, projects, and activities can be found at neherlab.org.
References and additional information
- Real-time tracking of seasonal influenza at nextflu.org
- Lab website: neherlab.org
- Existing code base: github.com/nextstrain
- Richard A. Neher and Trevor Bedford. Bioinformatics (Oxford, England), vol. 31, 3546--3548, 2015