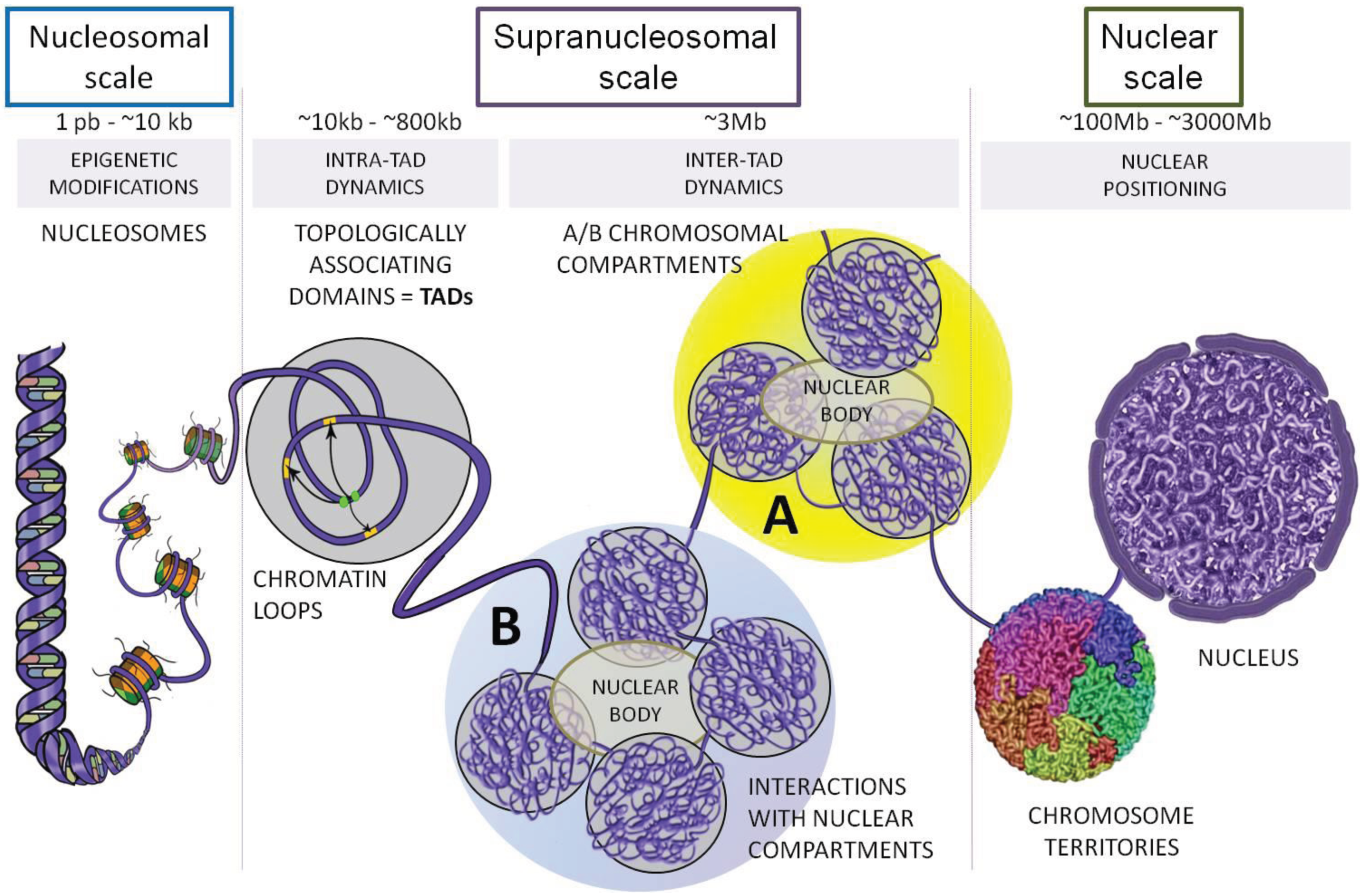
Packing a DNA into the nucleus while maintaining it accessible is a difficult task. Without any compactification the size of a DNA coil is \(\sim \sqrt{l_p L}\) where \(l_p=150bp = 50nm\) is the persistence length of DNA. Given a typical chromosome size of 100Mb corresponding to \(L=3\times 10^7 nm\), we would expect a typical end-to-end distance of \(\sqrt{10^9nm^2}\approx 30\mu m\). Eukaryotic cells achieve this major compactification by wrapping DNA around histone octamers that are subsequently packed into higher order structures.
 Vuthy Ea, Marie-Odile Baudement, Annick Lesne and Thierry Forné. Genes 2015, 6(3), 734-750
Vuthy Ea, Marie-Odile Baudement, Annick Lesne and Thierry Forné. Genes 2015, 6(3), 734-750
The complex of histone octamer and the DNA is called nucleosome. About 150bp (one persistence length!) is wrapped about 1.7 times around an octamer of 2 copies of H2A, H2B, H3 and H4 each. DNA is strongly bent in this configuration, which is maintained by many protein-DNA interactions.
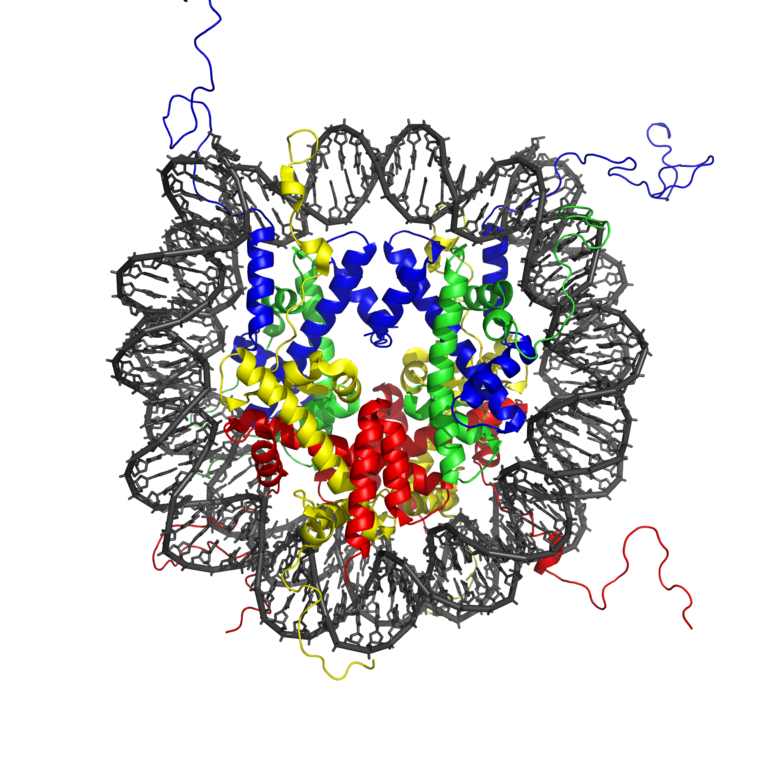
This extremely tight association between DNA and nucleosomes begs the question how DNA is kept accessible to regulatory proteins such as transcription factors. The last two decades have seen tremendous progress in figuring out how chromatin is maintained and remodeled. These mechanisms involved many different modification of histones (histone acetylation) and DNA (methylation). Furthermore, different variants of the histone proteins can be substituted for each other.
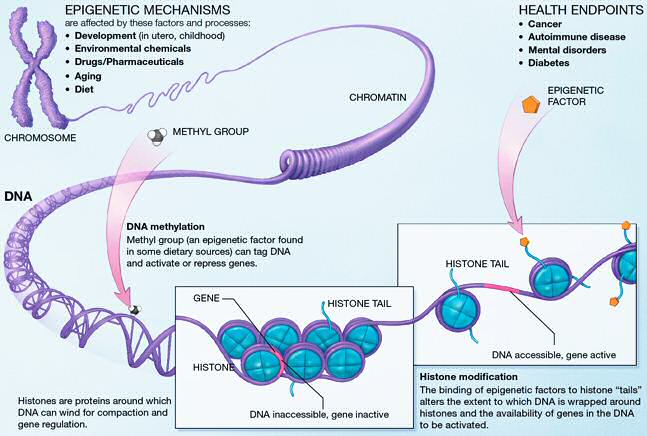
One aspect of epigenetic regulation is the positioning of histones on the DNA. Using deep sequencing techniques, it is now possible to determine the distribution of nucleosomes along the entire genome.
 Cizhong Jiang and B. Franklin Pugh, Nature Reviews Genetics
Cizhong Jiang and B. Franklin Pugh, Nature Reviews Genetics
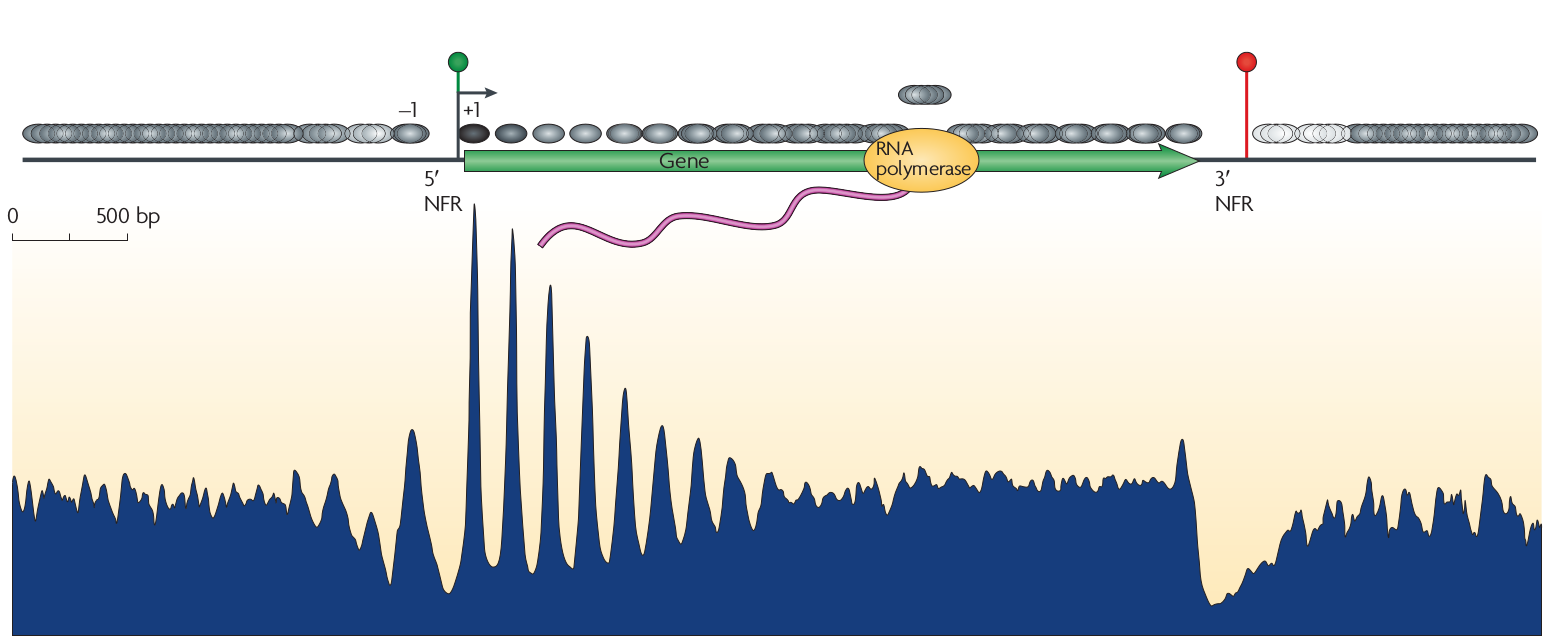
Nucleosomes have a preference for certain sequences (those that are particularly flexible at particular positions to facilitate wrapping around the histone octamer) and avoid others. These sequence preference allow explicit positioning of nucleosomes and it has been observed that transcription start sites tend to be nucleosome depleted. The upstream and down stream regions next the TSS tend to have periodic nucleosome occupancy. The degree to which these periodic patterns are explicitly positioned is not immediately clear. However, statistical modeling by Möbius and Gerland shows that a simple model of randomly placed nucleosomes next to a rigidly organized TSS is enough to explain these patterns (Tonks gas). The explicit comparison between the model and the data is shown below.
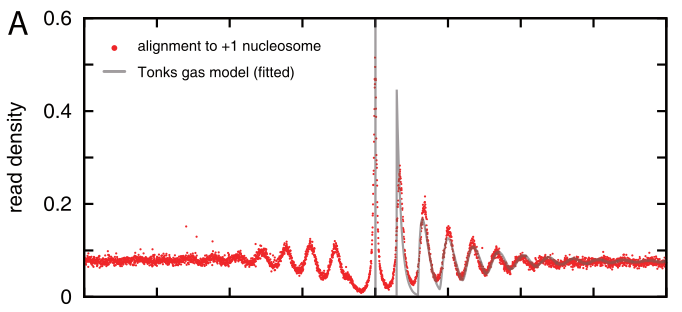
Density of reads mapping to nucleosomes in yeast compared to the prediction of the simple Tonks-gas model.
The discrete contributions of nucleosomes as distance 1,2,3,4 etc are shown separately in the plot below
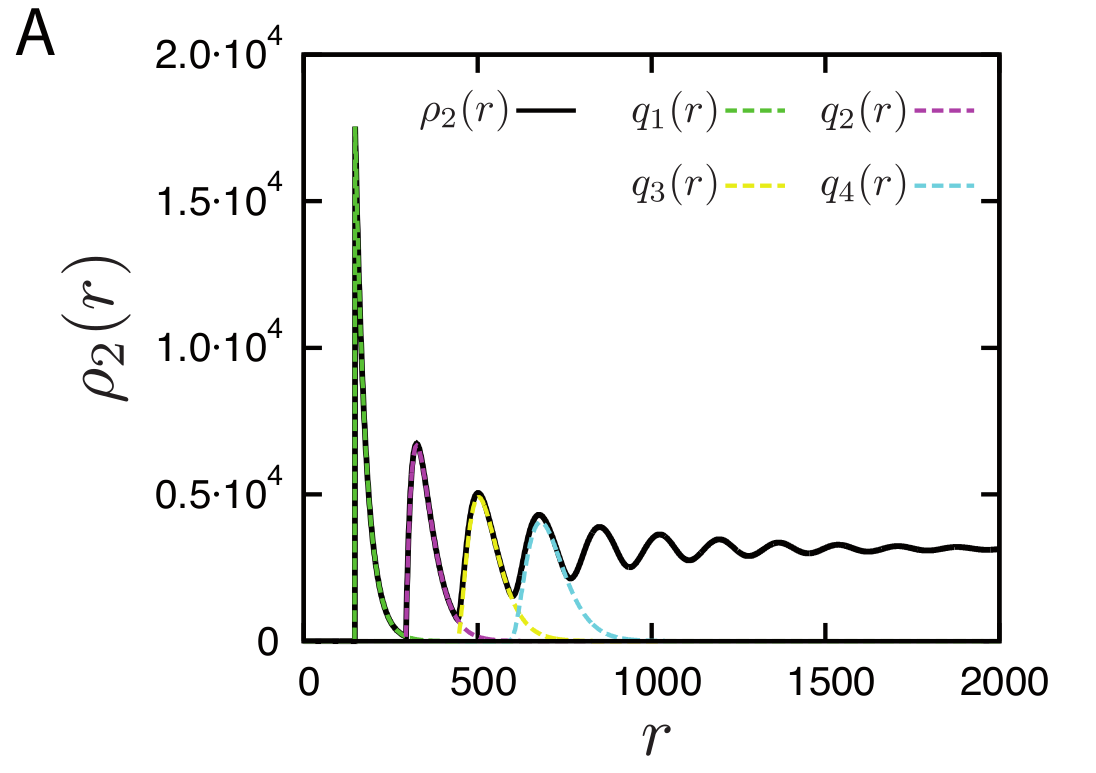
Density of reads mapping to nucleosomes in yeast compared to the prediction of the simple Tonks-gas model. With increasing distance, the peaks get broader and broader and eventually give rise to a uniform distribution.
Large scale chromosome organization
Beyond the microscopic organization of chromatin on the nucleosome scale, chromatin is organized in specific structures all the way to whole chromosome scales. The earliest indications of this were banding patterns in polytene chromosomes in Drosophila. Fluorescent probes can be used to monitor the location of particular loci within the condensed or open chromatin and multi-color labeling can reveal the relative location of several such loci. Recently, however, high throughput methods that can survey billions of interactions have enabled a much more detailed view of the 3D organization of chromatin.
3C, 4C, 5C, Hi-C: methods for characterizing chromatin
These new high-throughput methods are based on fixation of cross-links, ligation of pieces in physical proximity, and deep sequencing. The methods are derivatives of the original 3C (chromosome conformation capture). While 3C and 4C were restricted to specific or few locus interaction assays, HiC can be used to detect interactions across chromosomes.
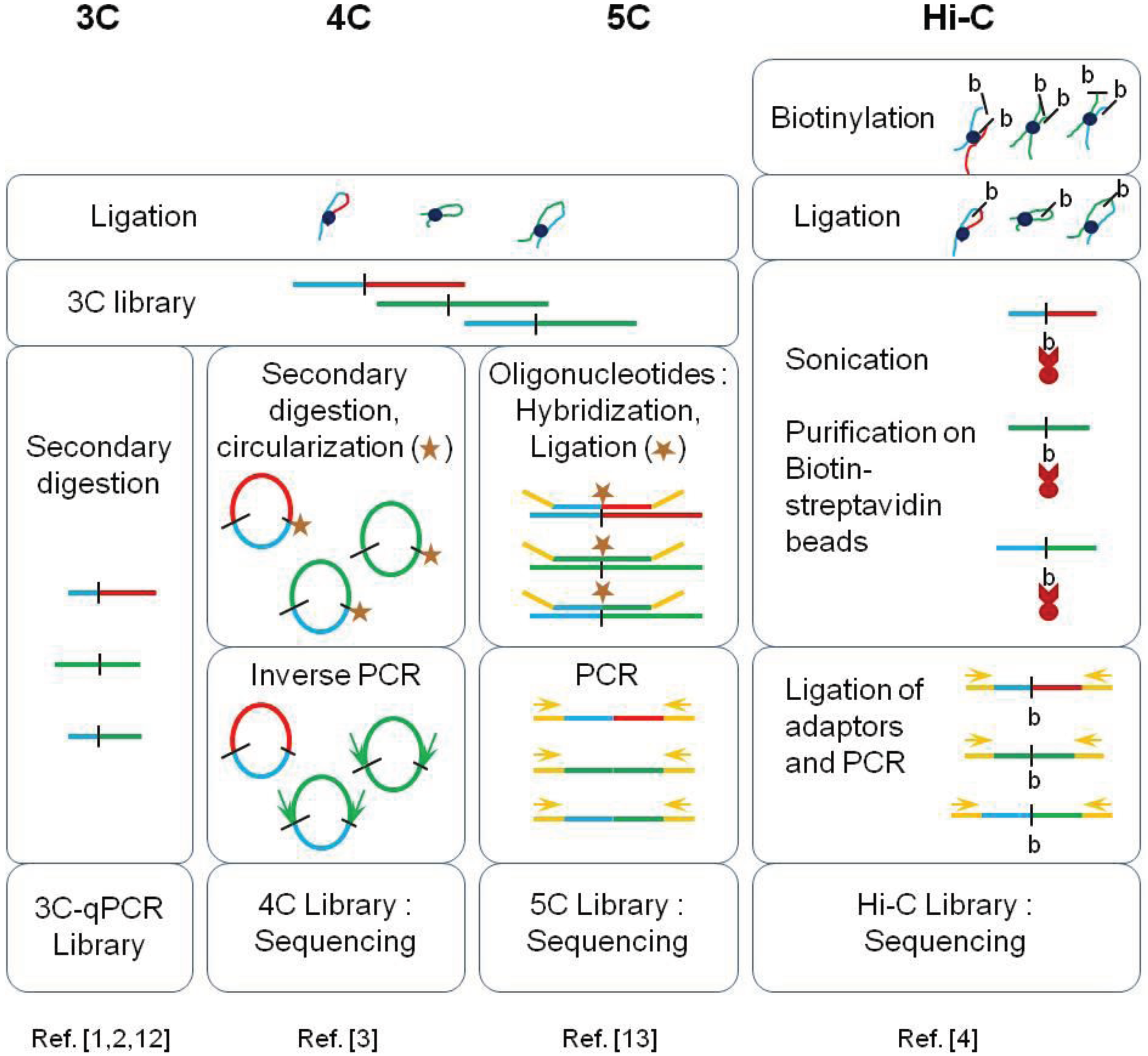 Vuthy Ea, Marie-Odile Baudement, Annick Lesne and Thierry Forné. Genes 2015, 6(3), 734-750
Vuthy Ea, Marie-Odile Baudement, Annick Lesne and Thierry Forné. Genes 2015, 6(3), 734-750
These methods generate detailed maps of how likely it is that two points on the chromosome touch.
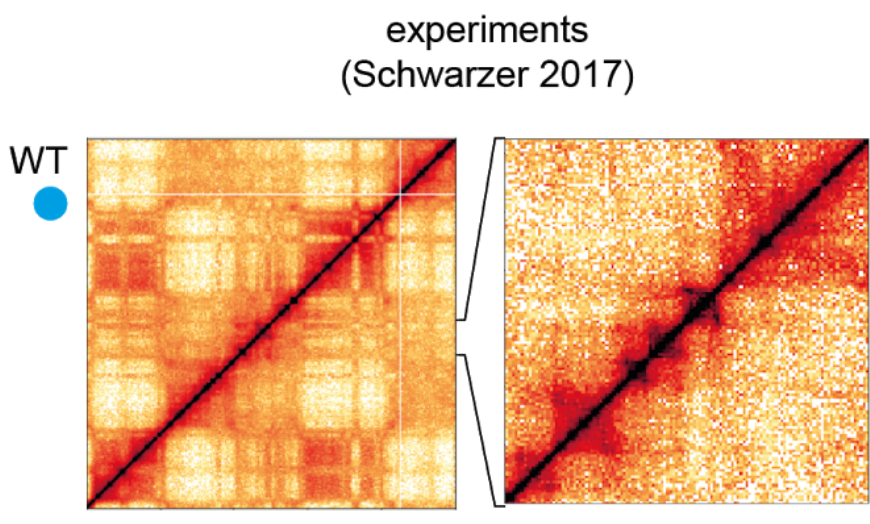
Example of a HiC map on chromosome 6. This shows an 50Mb interval. From Nuebler et al
Using HiC methods, scientists have shown that chromatin is organized hierarchically on multiple scales. At the largest scale, there are two domains (termed A/B). This two-domain structure is evident in the checkerboard pattern of HiC maps as seen in the top left figure below. These are segments that tend to interact with themselves and their next-nearest neighbor, but not their immediate neighbor.
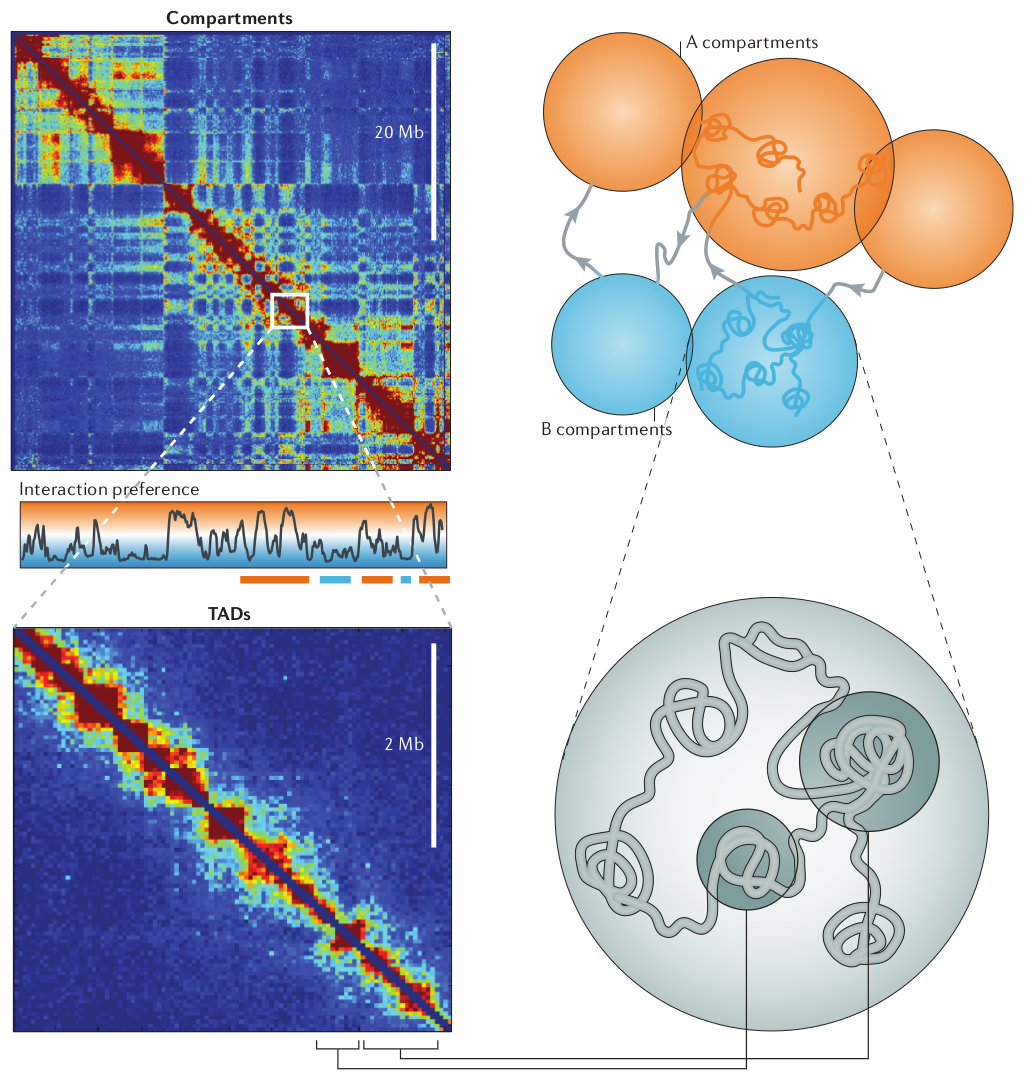
Deckker, Marti-Remon, Mirny. Nature Review Genetics, 2013
At finer scales, chromosome are organized into so called Topologically Associated Domains (short TADs). This really is just a fancy name of a local blob where the DNA tends to touches itself. We'll touch on how these TADs form and how they are maintained below.
For a more statistical characterization of the chromosome structure, it is instructive to calculate the probability that two points in the DNA interact as a function of distance. We have discussed explicit predictions for this probability in the context of polymer conformations where the dominant behavior for a random coil was \(s^{-3/2}\). This probability of touching decays much less rapidly in the nucleus.
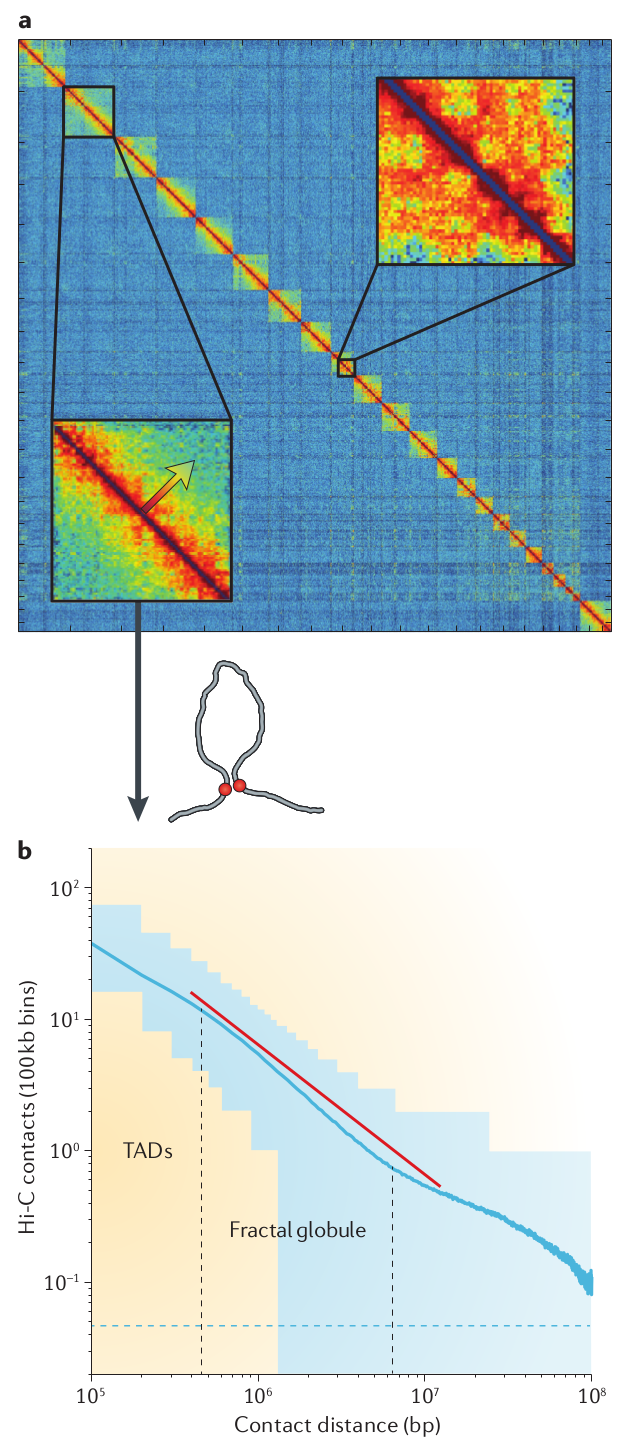
There is a fabulous tool to interrogate and explore chromosome conformation structure. It provides Google Maps like views into HiC data sets at every level and is available at higlass.io
Mechanisms of maintaining chromosome structure
This is a very recent field and some of the results are still very much in flux. The boundaries of TADs tend to be associated with a protein called CTCF. Leonid Mirny's group at MITA has put forward a hypothesis that TADs are maintained by a loop extrusion mechanisms. This model assumes that a certain type of ring-like motor protein binds on DNA and pulls the double strand through the ring until it hits a boundary defined by CTCF.
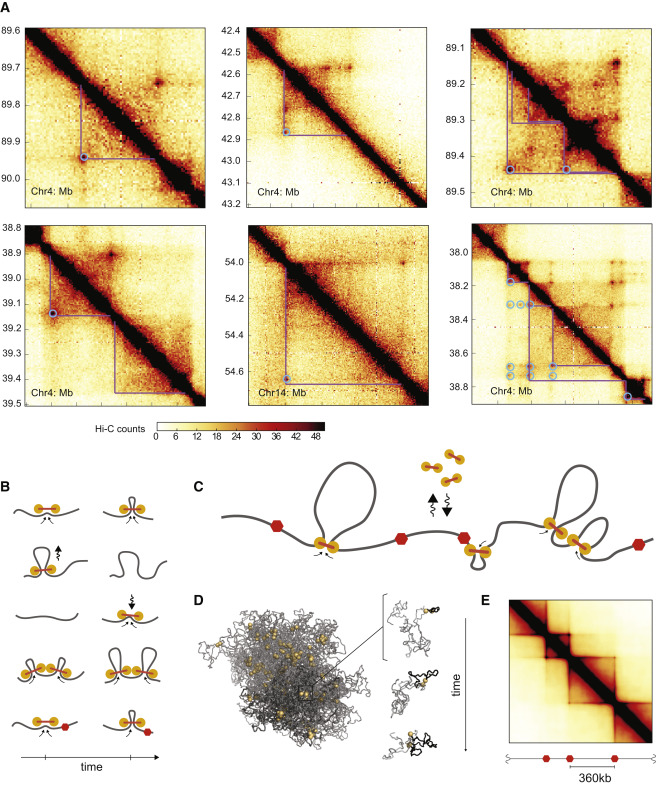
{kind=link}
Currently, the best candidate for this molecular motor are SMC cohesins.