About two month ago, Harris et al put up a preprint calling into question the relevance of soft-sweeps in the recent evolution in humans, flies and HIV. This preprint has generated a substantial amount of controversy and authors that were criticized by Harris et al felt they had to respond and defend their work. Alison Feder et al's response is a careful rebuttal of the critique by Harris et al. and there is little specific that I can add to it -- instead of looking at individual points of contention, I want to share here a more general take on this debate.
In summary: In RNA viruses, evidence for soft sweeps is overwhelming and a debate about whether sweeps are soft or hard is a waste of time. Keeping this debate alive by using simplified models and wrong assumptions for fundamental parameters of evolution is not helping. There are many much more interesting questions people should focus on.
Softsweeps are common in RNA viruses
HIV is one of the best examples of a rapidly adapting population. The co-evolution of HIV with the host immune system and its response to (suboptimal) drug treatment has been studied in detail at the genetic and phenotypic level in densely sampled time series. Similarly, seasonal influenza viruses evade the build-up of immunity in the human population by rapidly changing their surface proteins. In both of these viruses, softsweeps and parallel adaptation are a dominant mode of adaptation.
The fact that parallel adaptation is common in these populations is not surprising (unless one insists on unrealistically small population sizes, see below) and has been the null hypothesis of virologists for decades. The figure below shows trees of HA segments from influenza virus A(H3N2) and A(H1N1pdm) viruses isolated in the last two years. In both cases, a particular adaptation has risen to high frequency on different haplotypes.
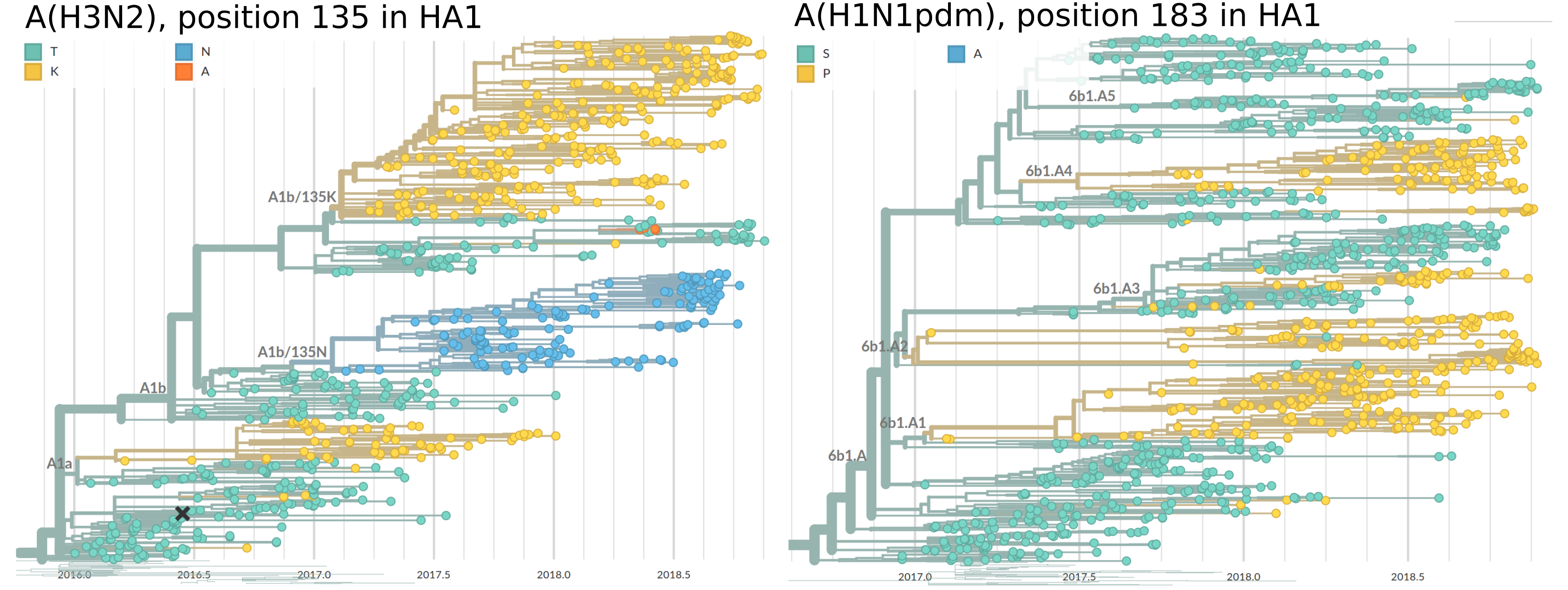 These screenshots from nexstrain show trees colored by amino acids at codon 135 (right) and 183 (left).
In both cases, several independent substitutions at these positions are ancestral to major circulating clades.
Parallel adaptation of this sort is the rule rather than an exception in seasonal influenza virus populations -- even though one of the competing lineages will eventually win and drive others extinct.
These screenshots from nexstrain show trees colored by amino acids at codon 135 (right) and 183 (left).
In both cases, several independent substitutions at these positions are ancestral to major circulating clades.
Parallel adaptation of this sort is the rule rather than an exception in seasonal influenza virus populations -- even though one of the competing lineages will eventually win and drive others extinct.
Similar examples are commonly observed in HIV. Most strikingly, immune escape early in infection often proceeds via dozens of distinct variants that differ at some position in the targeted epitope or the neighboring processing sites. The example below is from an early deep sequencing study by Fischer et al.
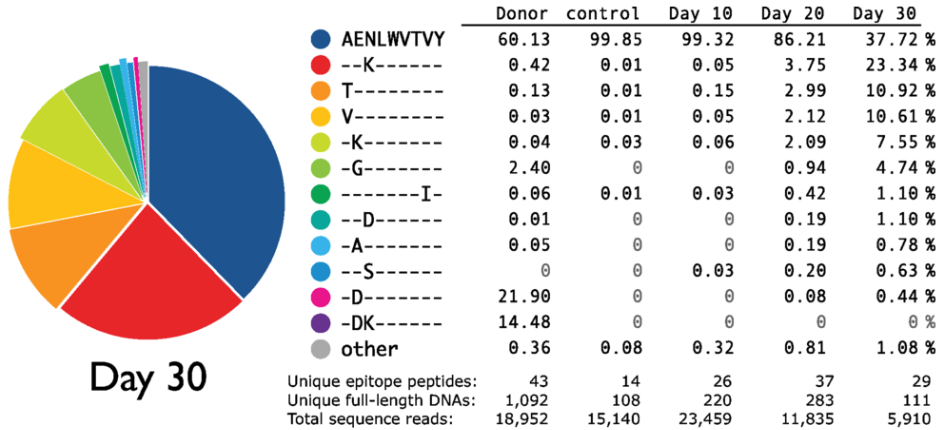 At day 30, the founder variant accounts for 38% of the sequence reads.
The remainder is composed of five variants above 3% and several rare more competing escape mutants.
In a similar fashion, drug resistance under mono-therapy typically emerges multiple times on different haplotypes -- often exploring different codons that code for the same amino acid.
At day 30, the founder variant accounts for 38% of the sequence reads.
The remainder is composed of five variants above 3% and several rare more competing escape mutants.
In a similar fashion, drug resistance under mono-therapy typically emerges multiple times on different haplotypes -- often exploring different codons that code for the same amino acid.
Multiple independent origins of an adaptation can either (i) root in diversity that existed before selective pressure was applied or (ii) arise in the declining population after the selection started. The relevant contributions of these two modes depend on the fitness effects of the mutations before and after conditions changed and distinguishing these two modes tends to be quite fuzzy. At the great majority of positions, however, we expect minor variants to pre-exist.
In fact, minor variation can be used to estimate the fitness costs of mutations at a large fraction of all positions in the HIV genome suggesting an approximate mutation/selection balance (see Zanini et al and Theys et al). This clearly implies that most mutations pre-exist and are readily selected for when drug treatment or immune selection turn them into beneficial mutations. Hence adaptation from standing variation is plausible for all but the most deleterious mutations.
Why are we even having this debate?
Viral populations are large (10s of millions of cells are infected every day within a chronically infected HIV patient, there are millions of influenza cases a day) and mutation rates are on the order of \(10^{-5}\) per site and day. Hence every mutation is expected to be produced hundreds of times every day.
Why do Harris et al then assume an HIV population size of \(10^4\) and therapy induced bottlenecks of size 1000, 100, and 10 (as well as way too small selection coefficients)? Population geneticists are used to estimating population sizes from the levels of genetic diversity that are maintained in the population. Neutral genetic diversity is proportional to the product of the time to the most recent common ancestor \(T_{mrca}\) and the mutation rate \(\mu\). In the simplest neutral models of population genetics \(T_{mrca}\sim N\) and this equivalence is used to estimated the 'effective population size' \(N_e\) from diversity via \(N_e \sim T_{mrca}/\mu\).
In HIV, the genetic diversity tends to saturate at a pairwise distances of about one percent suggesting \(T_{mrca}\sim 10^3-10^4\) (after accounting for conserved sites etc). In most microbial populations, however, there is no reason to believe that \(T_{mrca}\) has anything to do with size of the population that is exploring beneficial mutations -- most certainly not in HIV or influenza virus populations. Taking estimates of \(N_e\) based on genetic diversity at face value will result in erroneous conclusions. Genetic diversity in rapidly adapting populations is controlled by selection, both positive and negative, rather than drift and the population size exploring mutations is orders of magnitudes larger than the number of generations to the most recent common ancestor. Population size can even by anticorrelated with diversity: Influenza virus A(H3N2) has the largest population size of seasonal influenza viruses (as measured by prevalence), but has historically had lowest diversity.
We don't even need to rely on theoretical arguments here. The size of the HIV population that contributes beneficial drug resistance mutations has been estimated in animal models. Boltz et al for example find that at least \(10^6\) infected cells contribute to future generations in HIV infected macaques. They measure mutation frequencies down to \(10^{-5}\) and show that some resistance mutations (M184I, K65R) are found as rare variants before treatment. In contrast, K103N was not detected above \(10^{-5}\) prior to treatment, but multiple codons (AAC and AAT) coding for Asparagine at position 103 were observed a few days after treatment despite a transient 100fold reduction in viral load. Hence even when a mutation is deleterious enough not to be observed above frequencies \(10^{-5}\) (suggesting costs of order one) and the population size is rapidly declining due to drug treatment, the viral population is still large enough to discover and amplify this resistance mutations many times.
Viral genetic diversity data still holds many mysteries
...but whether sweeps are soft or hard is not one of them. Most available evidence suggests that selection and the environment, rather than the discovery of particular mutations, determine diversity and adaptation. This selective landscape, however, is not well understood. We should strive to understand how an environment that varies in time and space drives virus adaptation, how viruses interact with diverse immune system(s), and how biophysical constraints of viral proteins limit the space of possibilities. Densely sampled human pathogens are unique in that extensive data on prevalence history, serology, and the evolutionary dynamics of different variants are available. They are one of the most promising systems to unravel the interactions that drive rapid adaptation. Answers to these question are key to quantitatively understand which factors determine the distinct behaviors of the different seasonal influenza virus lineages, or why other respiratory viruses like rhino- or enteroviruses have very different evolutionary patterns.
Bigger organisms and larger genomes
Much of the discussion above focused on RNA viruses -- I know the literature on them much better than that on organisms with larger genomes. The latter tend to have much lower mutation rates, making it harder for a population to sample the mutational space and hence select for the same mutation on different backgrounds. However, what these populations lack in mutation rate, they often make up for by larger mutational targets or different modes of adaptation. Bacteria for example adapt by picking up genes from the environment and resistance genes spread on diverse background across species boundaries. Furthermore, their populations are easily big enough to explore all single point mutations even at mutation rate of \(10^{-9}\) per site and generation.
Similar arguments hold for small animals like fruit flies that with their enormous populations can sample single point mutations exhaustively, see Karasov et al. If these mutations are strongly adaptive, they will sweep from multiple independent origins. Deviations from panmixia can help to keep these independent and possibly competing adaptations around for a while.
Conclusion
To quote Alison, "something that likely happens at least once likely happens multiple times". As a corollary, populations that regularly need to change specific loci in their genomes to survive are unlikely to be mutation limited and will largely adapt via softsweeps tracking a changing environment rather than waiting for rare lucky mutations. That does not mean that unique "hard-sweep" like events don't happen -- only that they are atypical and by definition rare. If environmental challenges that require such adaptations were frequent, the population would go extinct. Many crucial transitions in the history of most species will have been such rare "hard sweeps", e.g. a host jump of a virus. But there is no contradiction here: every day business in a gradually changing environment proceeds mostly via softsweeps, while major transitions are rare and plausibly of single origin.
In fact combination drug therapy for HIV is precisely set-up to engineer such a dramatic change of environment: Several drugs are combined to erect a large genetic barrier. Resistance requires the simultaneous acquisition of multiple previously costly mutations that would drive the HIV population extinct if it wasn't for the latent reservoir.
While simple effective models might fit some data by adjusting phenomenological parameters like \(N_e\), their predictions typically conflict with other observations. Models parameterized to fit genetic diversity on the time scale of years will not give meaningful prediction on evolutionary processes that take place over a few days. Ignoring the realities of large rapidly adapting populations and modeling them as small neutral populations (for mathematical convenience or otherwise) is muddying the waters.