Nextstrain uses a program called 'auspice' to create the colourful and interactive webpages you probably associate with Nextstrain. We'll talk more about the details of auspice later, but for the moment, we'll show you how you can explore data using auspice, and use it to illustrate some of the concepts just covered.
To do this, we'll look at a small sub-sample of the Zika virus sequences used to create the Zika page on Nextstrain.
Follow this link to take a look at this 'Zika tutorial'.
The default view shows three main panels on your screen:
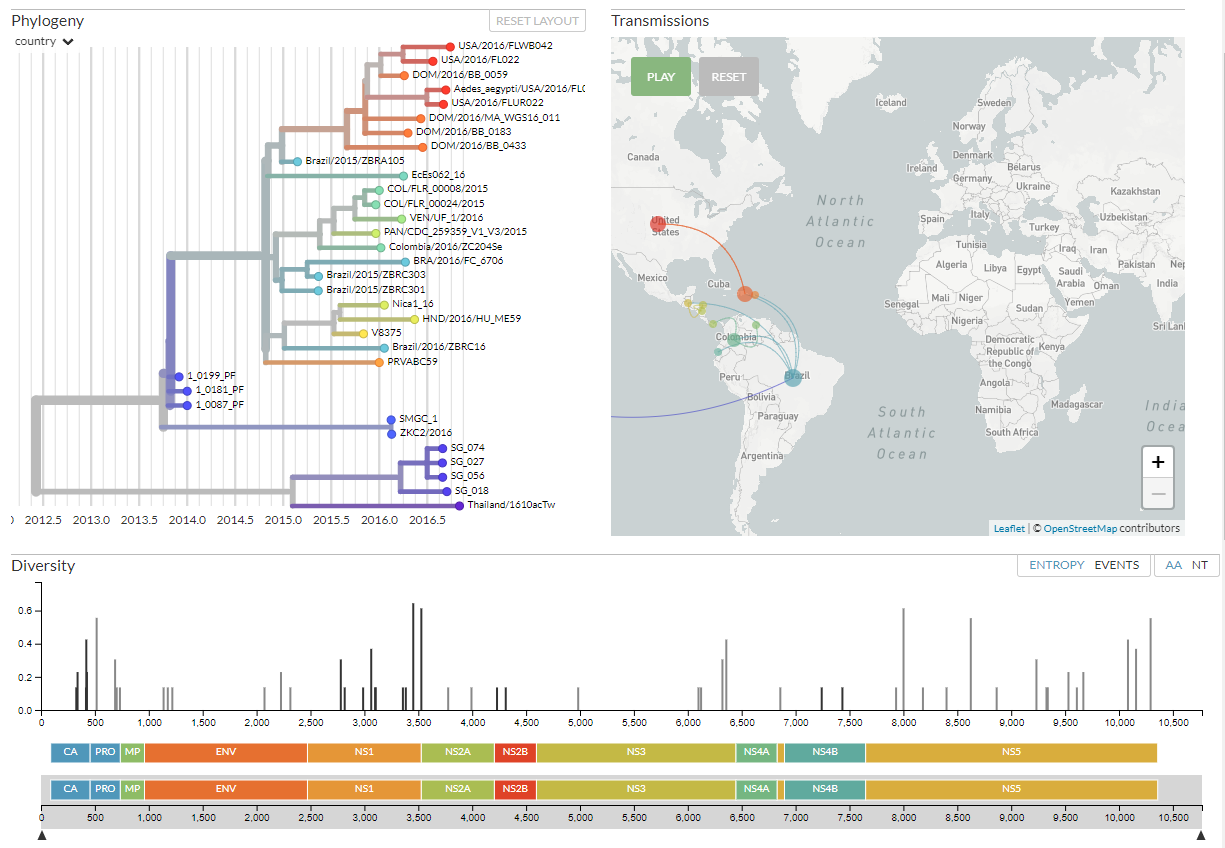
(If you don’t see all three panels at once, try making your browser larger, or scroll down to see them.)
In the top-left is the phylogeny, with the sequences used in the analysis. Top-right shows a map showing
where the sequences are from. Finally, at the bottom is a ‘diversity’ panel that shows the full Zika genome,
its genes, and mutations found in this analysis.
Let’s look more closely at each panel
Phylogeny Panel
In this dataset, the phylogeny is coloured by ‘country’ (country of sampling), and is displayed as a time-resolved tree. If you can’t see a legend explaining the colour of each country, click the small arrow next to ‘country’ in the top-left of the phylogeny panel.
You can see the x-axis is in years, and each sequence will be positioned horizontally according to sample date. If any undated samples had their dates estimated in the analysis, they’ll be positioned at their estimated date.
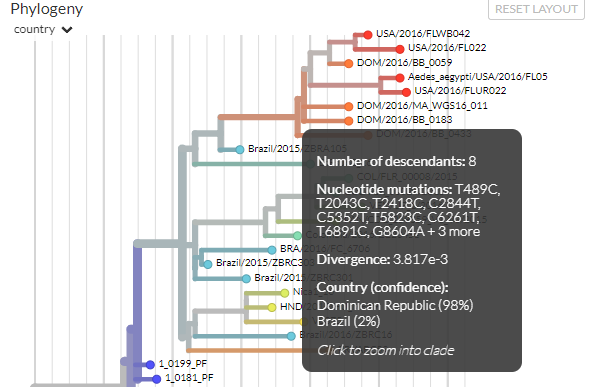
We can get more information about the phylogeny just by mousing over different parts of it. Hold your mouse (don’t click) over the large branch located almost in the middle of the panel:
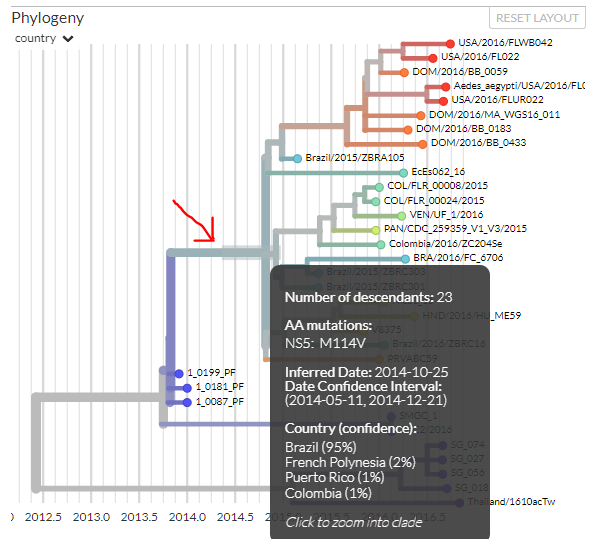
We can see that this branch has 23 descendants, and we can also see there was one amino-acid mutation that occurred on this branch and passed on to all its descendants (though further mutations at this site may have occurred in some descendants). This mutation was in the gene NS5; ‘M114V’ means that at codon position 114, a methionine (‘M’) changed to a valine (‘V’). We’ll explore mutations a little more in connection with the bottom panel.
The mouse-over text also shows us the estimated date for this branch, and confidence intervals for that estimate. Similarly, the branch is coloured grey-green, and we can see this is because the most likely location estimated is Brazil, which is greenish (the grey tinge comes from not being 100% confident in Brazil being the location).
Mouse over different branches in the tree and see how they differ. Mouse over some tips to show the info box -- can you tell which had sample dates, and which were estimated during analysis? If you click on tips, you can bring up more information about that sample.
Let’s take a closer look at part of the tree -- we can click on branches to zoom in. Click on the branch leading to the red sequences at the top:
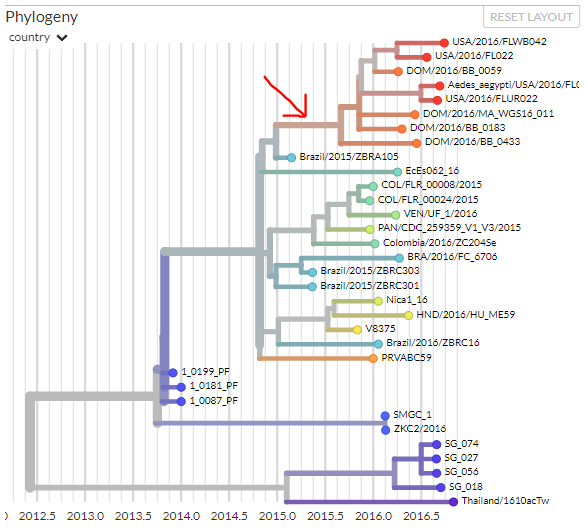
Since this tree is small, zooming in doesn’t change our view much, but in larger trees this can be very useful. If you look at the map and diversity panels, you’ll notice they’ve changed to reflect that we’re only looking at a sub-sample of the sequences now.
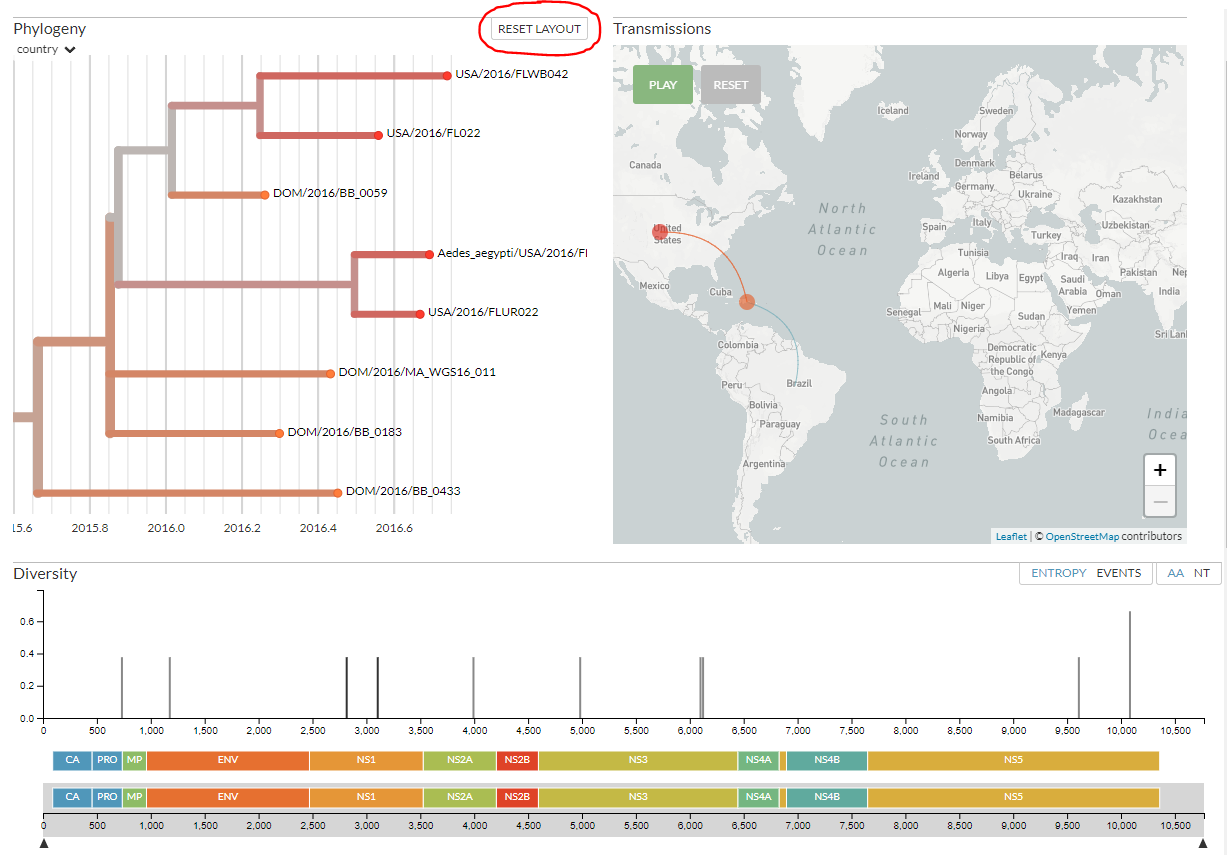
Click on ‘reset layout’ in the top-right of the Phylogeny panel (circled above) to go back to the full view. Click on a few other branches to zoom in, and see how the map and diversity panels adjust.
Control Panel
The control panel on the left can also change how the phylogeny is displayed. Using the sliders on either end of the ‘date range’, note how the panels respond as you change the range of dates that are viewable. (Put the sliders back to the ends afterwards, so the whole phylogeny is visible.)
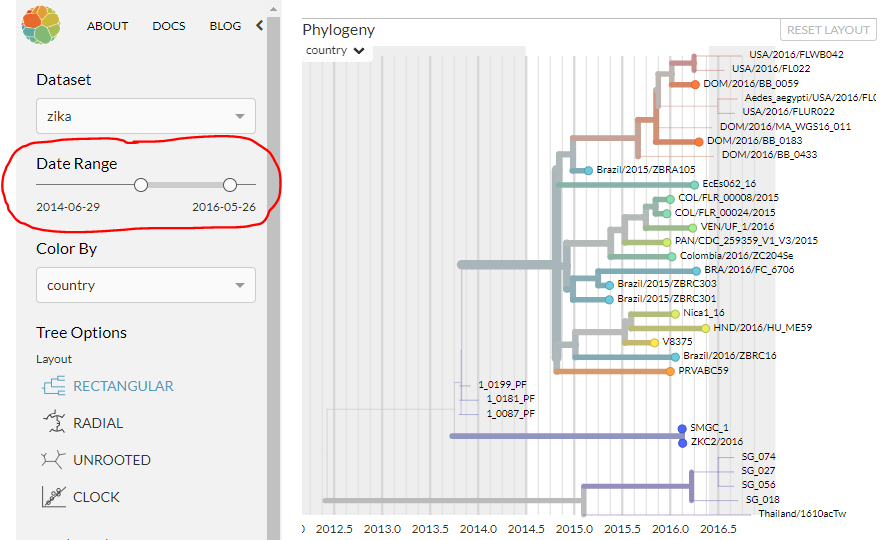
Next, using the ‘color by’ dropdown menu, try colouring the phylogeny by region, author, and date. We’ll come back to ‘genotype’ in the moment.
You can also change the layout of the phylogeny -- try clicking on ‘Radial’ and ‘Unrooted’ under ‘Tree Options - Layout’ and see how the tree changes.
Finally, click on the ‘Clock’ layout option -- it looks quite different:
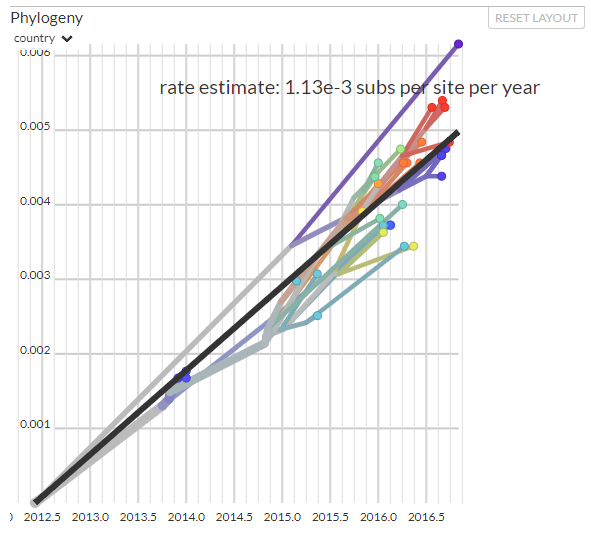
This view shows us divergence (number of mutations since the common ancestor) on the y-axis and the sampling date on the x-axis. In the ideal case, all points roughly fall on a line and the slope of this line is the evolutionary rate, that is the number of substitutions that accumulate per year.
Go back to ‘rectangular’ view, then click on ‘Divergence’ under ‘Branch Length’. The x-axis on the phylogeny is now shown with tips plotted according to their divergence (in substitutions) from the root, rather than by time. Mousing over branches and tips now will show the divergence, rather than the date.
Map Panel
The map panel is not as interactive as the phylogeny panel, but does change in to reflect actions like zooming or changing the dates on the phylogeny. Using the ‘play’ button in the top-left of the map panel, you can move through time on the phylogeny, and see how the virus may have spread geographically. How accurate these reconstructions are is dependent on many things, so it’s important to consider if showing such links is appropriate for your data.
You can adjust the speed of the animation using the ‘Map Options’ on the control panel. You can also change what level of location detail is shown on the map -- try changing the ‘Geographic resolution’ to ‘region’.
Diversity Panel
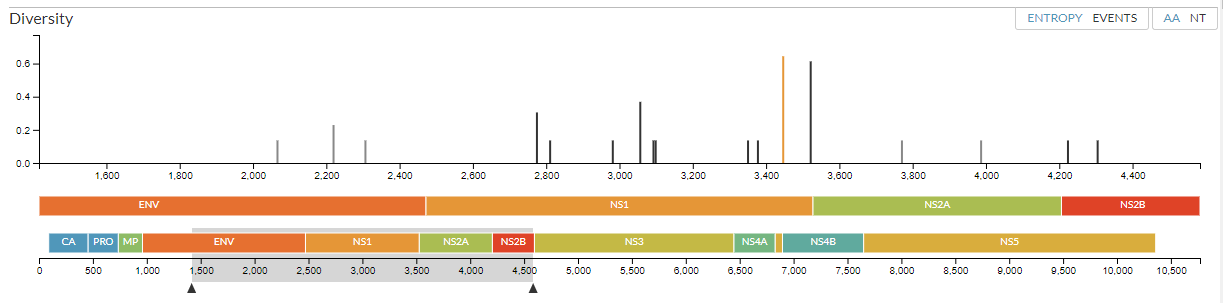
The diversity panel is a good way to look at mutations on the phylogeny. It allows you to spot regions in the genome that evolve more rapidly than others, for example because a particular region is targeted by the host immune system and changes quickly.
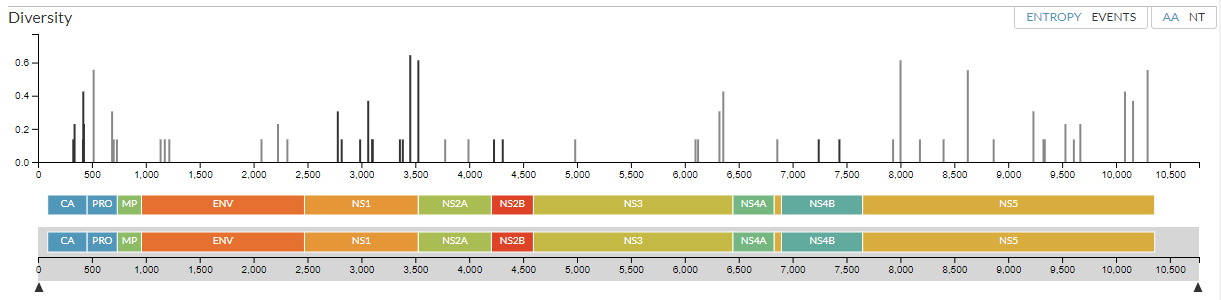
The top of the panel shows the a measure of how much an amino acid has changed across the tree. Each line represents a position that has changed, plotted along the genome to correspond with the two panels below.
If you’d rather look at the number of times an amino acid has changed, change from ‘entropy’ to ‘events’ in the top right of the panel -- notice the amino-acids all now have discrete y-values (here, either 1 or 2 changes). You can also look at nucleotide changes by switching from ‘AA’ to ‘NT’ in the top right of the panel. When you switch to ‘NT’, the information that appears when you mouse over the phylogeny will also change - it will now show nucleotide mutations for each branch.
Go back to ‘entropy’ ‘AA’ view. We can see exactly how an amino acid (or nucleotide) has changed across the tree by clicking on that location. Click on the site with the highest entropy:
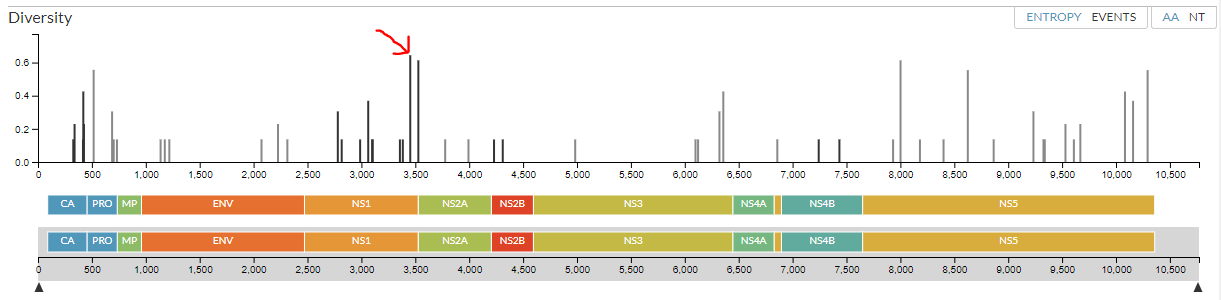
Notice this bar is now highlighted (it’s turned orange). The middle part of the diversity panel also looks different - it’s zoomed in so that the gene where this mutation has occurred (NS1) is centered. You can see the area that’s zoomed in by looking at the bottom part of the panel - the highlighted grey area shows the zoomed area on the middle section. The two arrows on either size of the grey area can be dragged to zoom in and out.
The phylogeny has changed as well. Click the small arrow next to ‘Genotype at NS1 site 324’ in the top-left of the phylogeny panel so that you can see the legend.
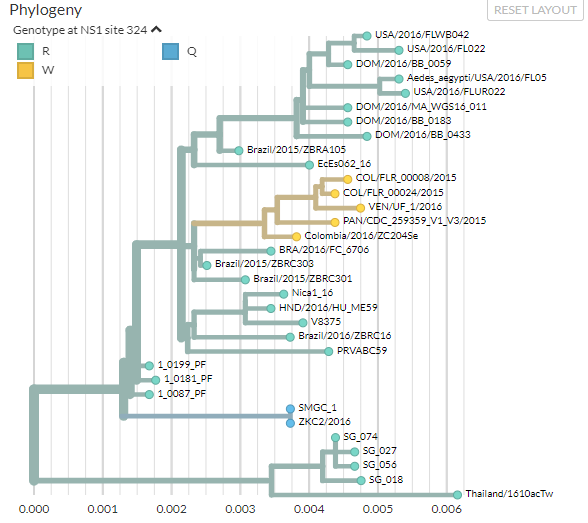
The phylogeny is now coloured by the amino acid present at position 324 in the NS1 gene. Most of the branches and tips have an arginine (‘R’), but some have mutated to tryptophan (‘W’) or glutamine (‘Q’).
Click on another site and see how the phylogeny changes. Switch to nucleotide ‘NT’ view, and click a few more sites -- the phylogeny will now show these mutations. Why are there more nucleotide mutations than amino acid mutations?
Zoom out in the diversity panel by dragging the black triangles to either end of the Zika genome, or by clicking outside of the zoomed area, when your cursor forms a cross. Go back to ‘entropy’ ‘AA’ view.
What if you’re interested in a particular gene or position? On the control panel, go to the ‘Color by’ drop-down menu and select ‘genotype’.
In the drop-down menu that appears directly below (which now shows ‘genotype’) select ‘NS5’. The diversity panel will zoom in to this gene so you can more clearly see the sites that have changed.
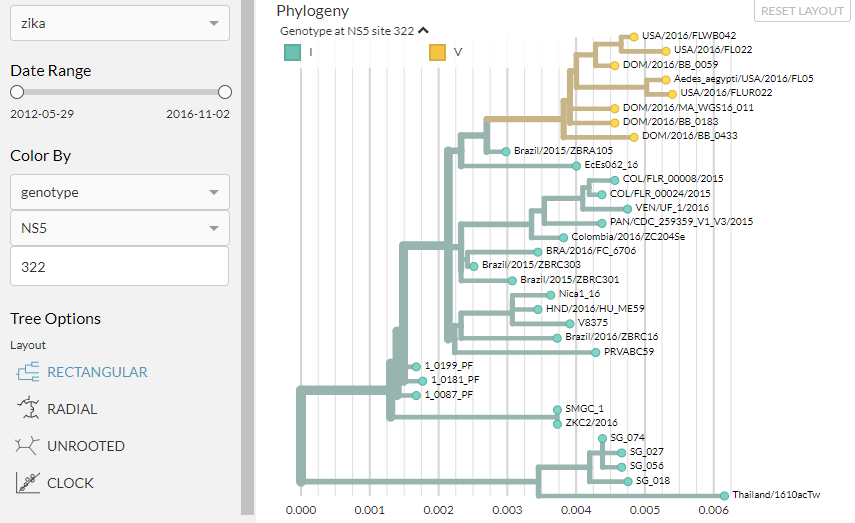
Mousing over the long branch leading to the top cluster on the phylogeny, we can see there were two mutations in NS5, at position 322 and 878:
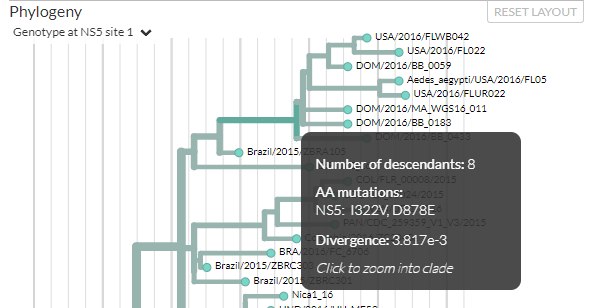
We can colour the phylogeny by these locations by typing ‘322’ into the box below where we selected ‘NS5’ in the control panel:
Now switch to ‘NT’ view in the diversity panel. Mousing over the same branch, you can see there are many more nucleotide mutations than amino acid mutations:
For nucleotide mutations, we can colour the phylogeny by multiple positions at once. As before, select ‘genotype’ under the ‘Color by’ dropdown. This time, leave the second dropdown box set as ‘nucleotide’. Type in the following positions, separated by commas but without spaces: 3501,3972,6966. Press enter, and ensure you can see the legend at the top of the phylogeny panel:
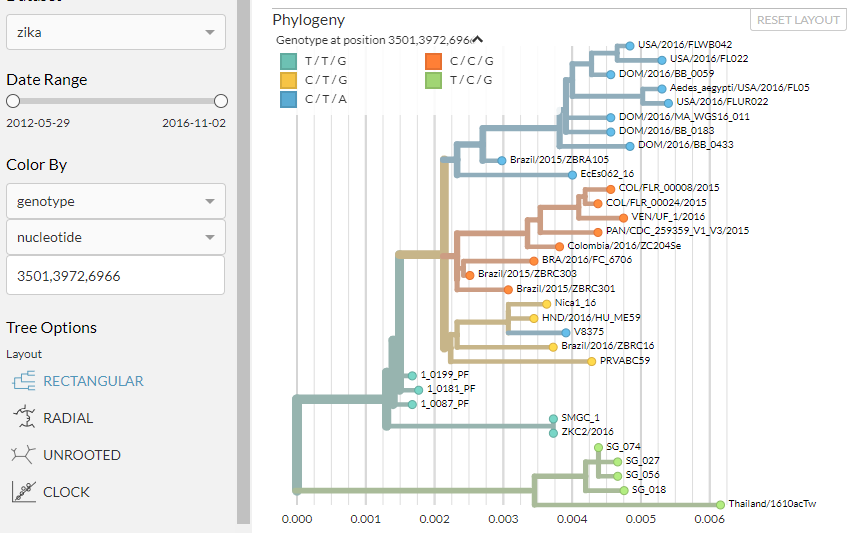
The phylogeny is now coloured by the nucleotide present at each of these three positions.