Analysing SARS-COV-2 data
This is the last part of the Nextstrain workshop. One can either try things with their own data or choose to focus on SARS-COV-2. This document provides some guidelines to the SARS-COV-2 analysis.
Questions
These are some suggestions concerning the questions you should try to answer using Nextstrain. Feel free to focus on other questions if you have something specific in mind. - How many clades is there in your data ? What do they correspond to? - How fast does SARS-COV-2 evolve overall? - Is the root of the tree correctly inferred? - Where are most of the mutations happening? What does this gene correspond to? - What are some of the mutations characterizing the different variants?
Downloading sequence dataset
Use online resources to create a SARS-COV-2 sequence dataset of around 1000 sequences. In our case we will be using NCBI virus resource to generate a suitable dataset.
You will arrive there
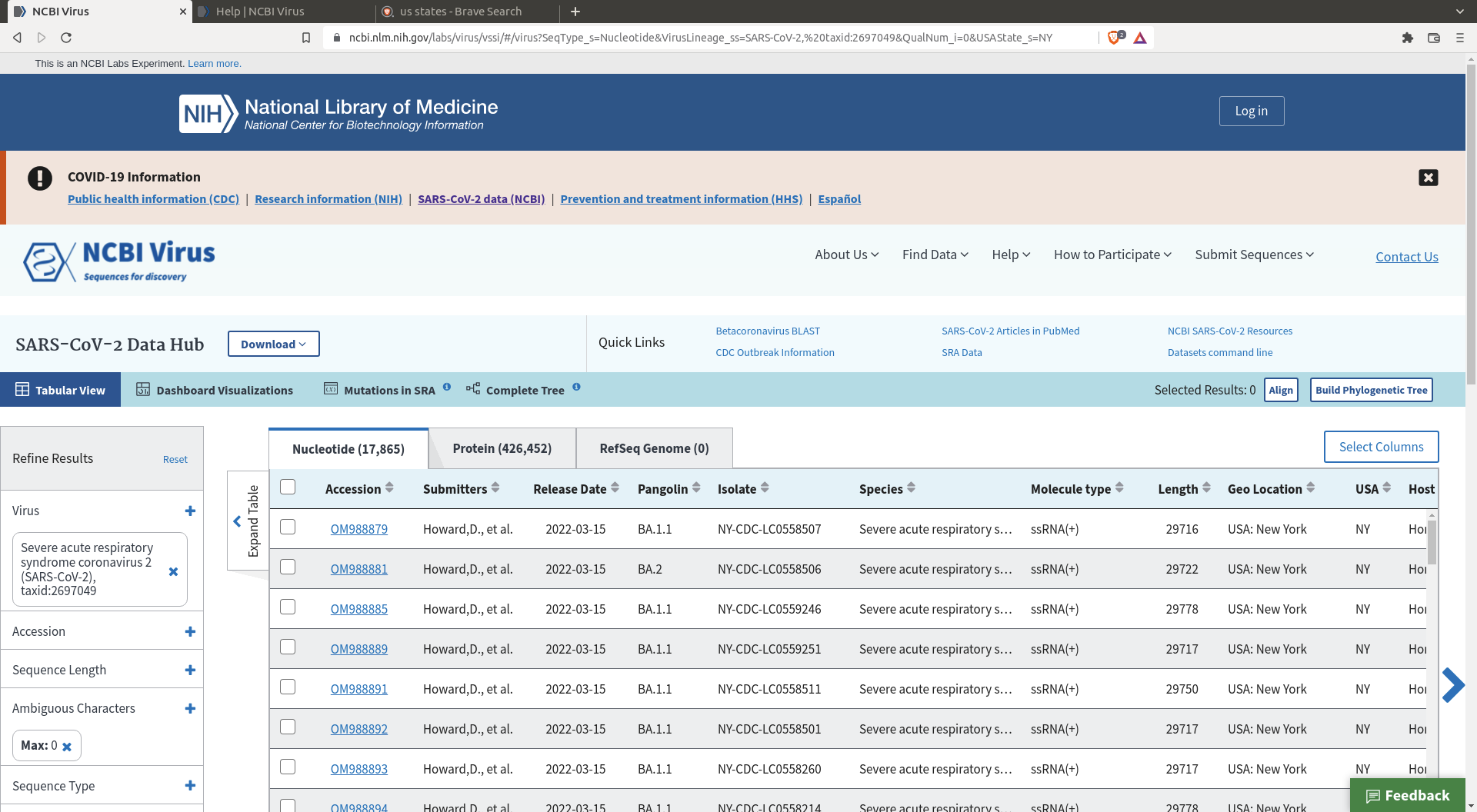
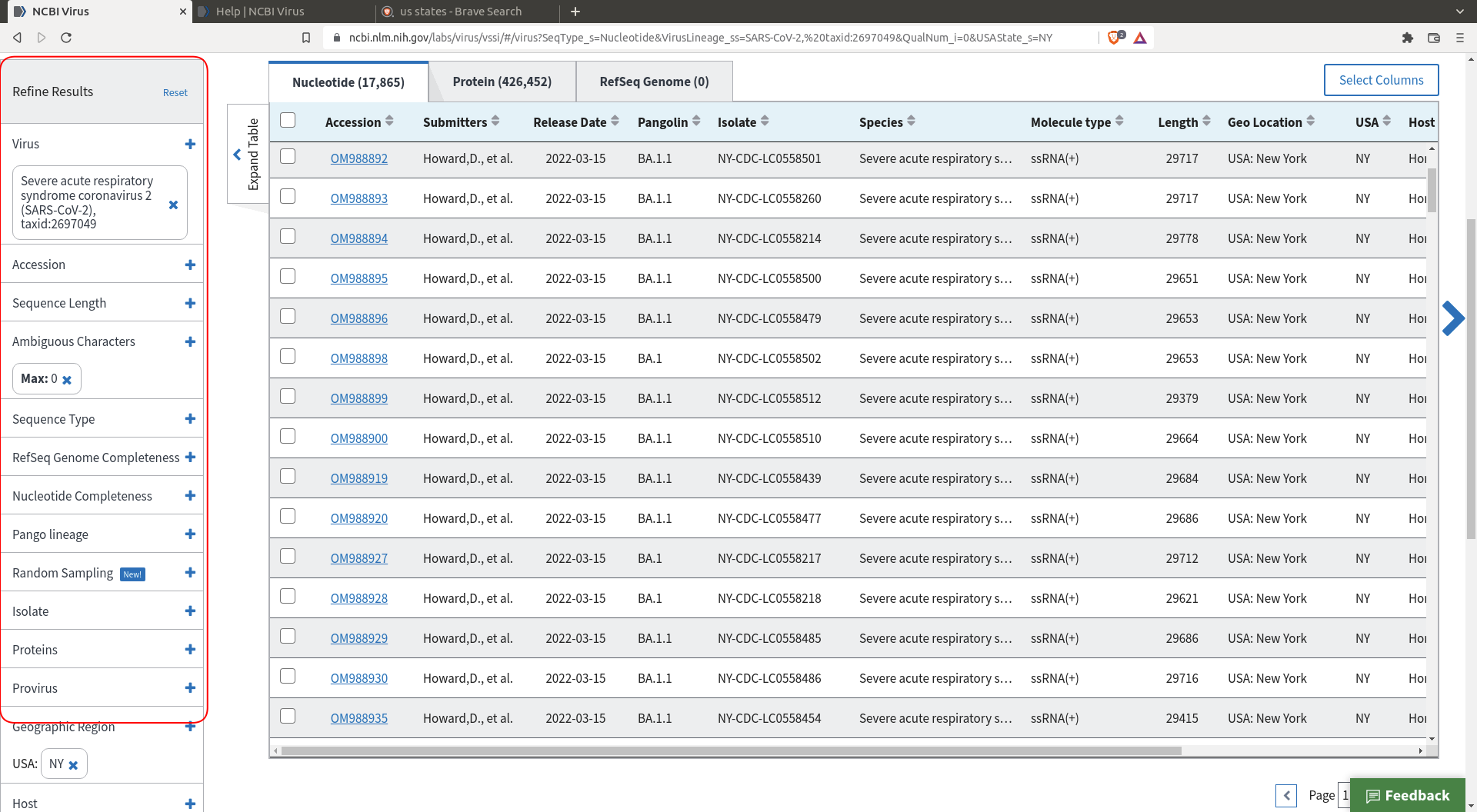
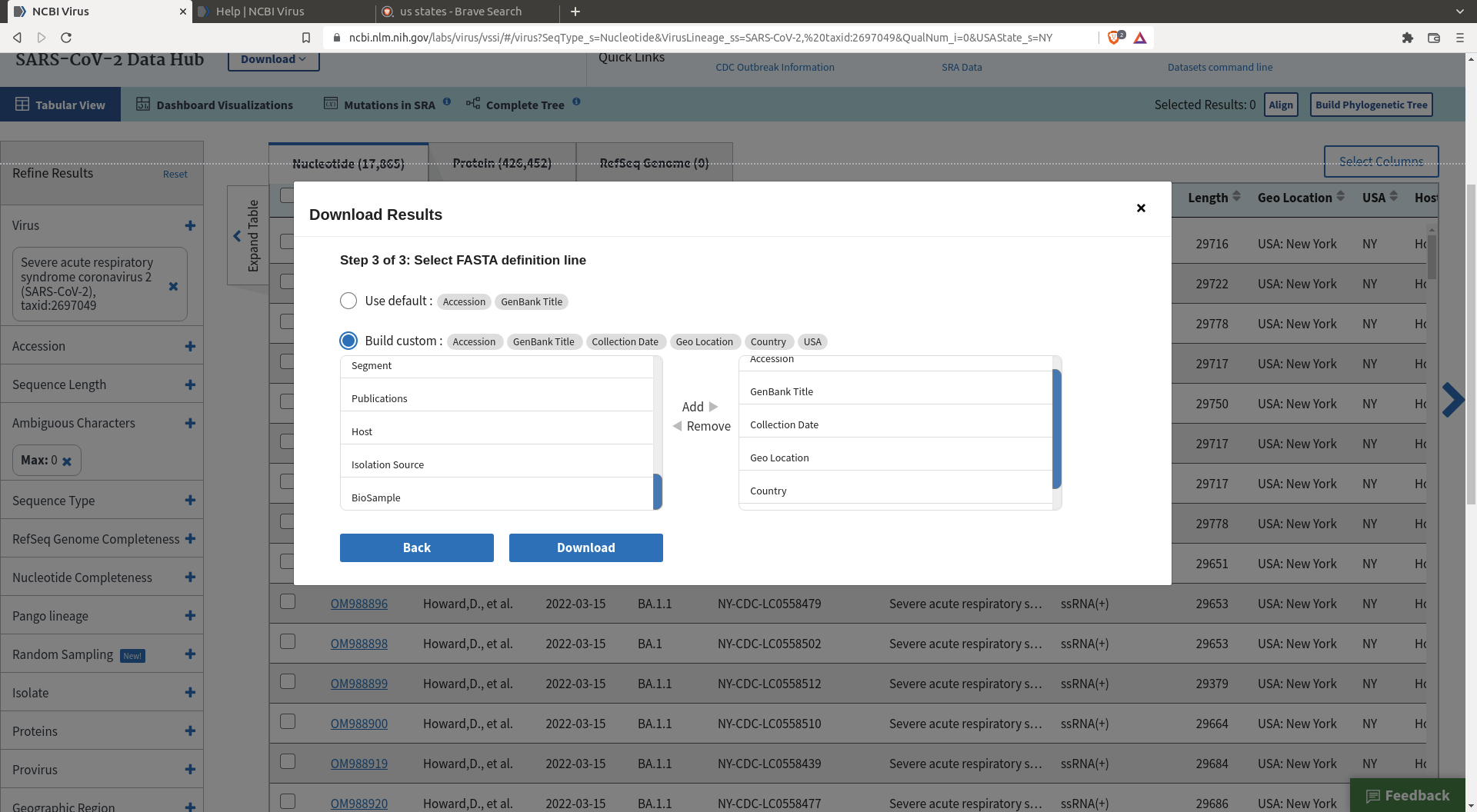
The interface allows you to download a random selection of the many sequences that are available. This will be helpful for our purpose as we only want to analyze a few thousand sequences, not a few millions. First, filter the sequences to a subset that best suits your questions
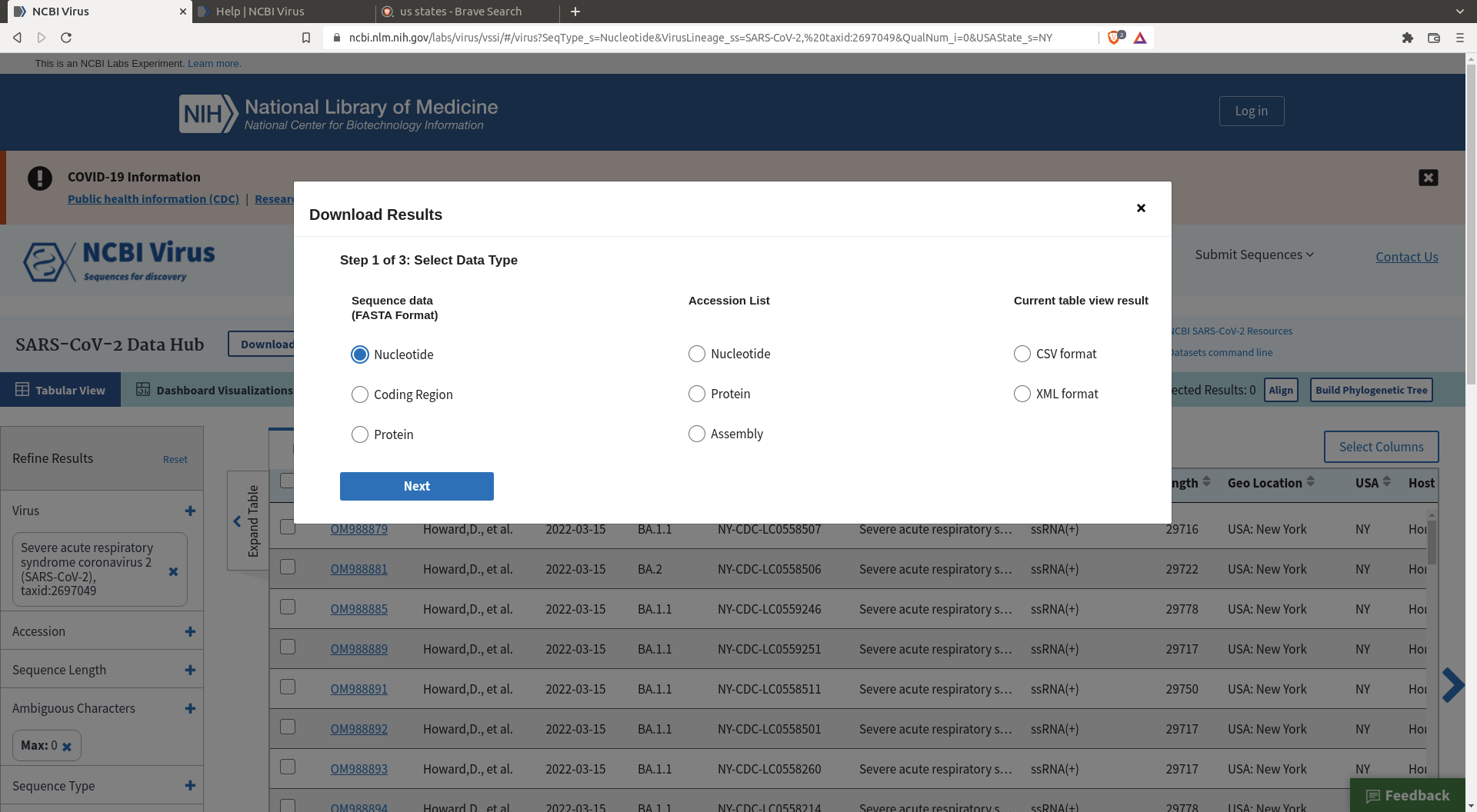
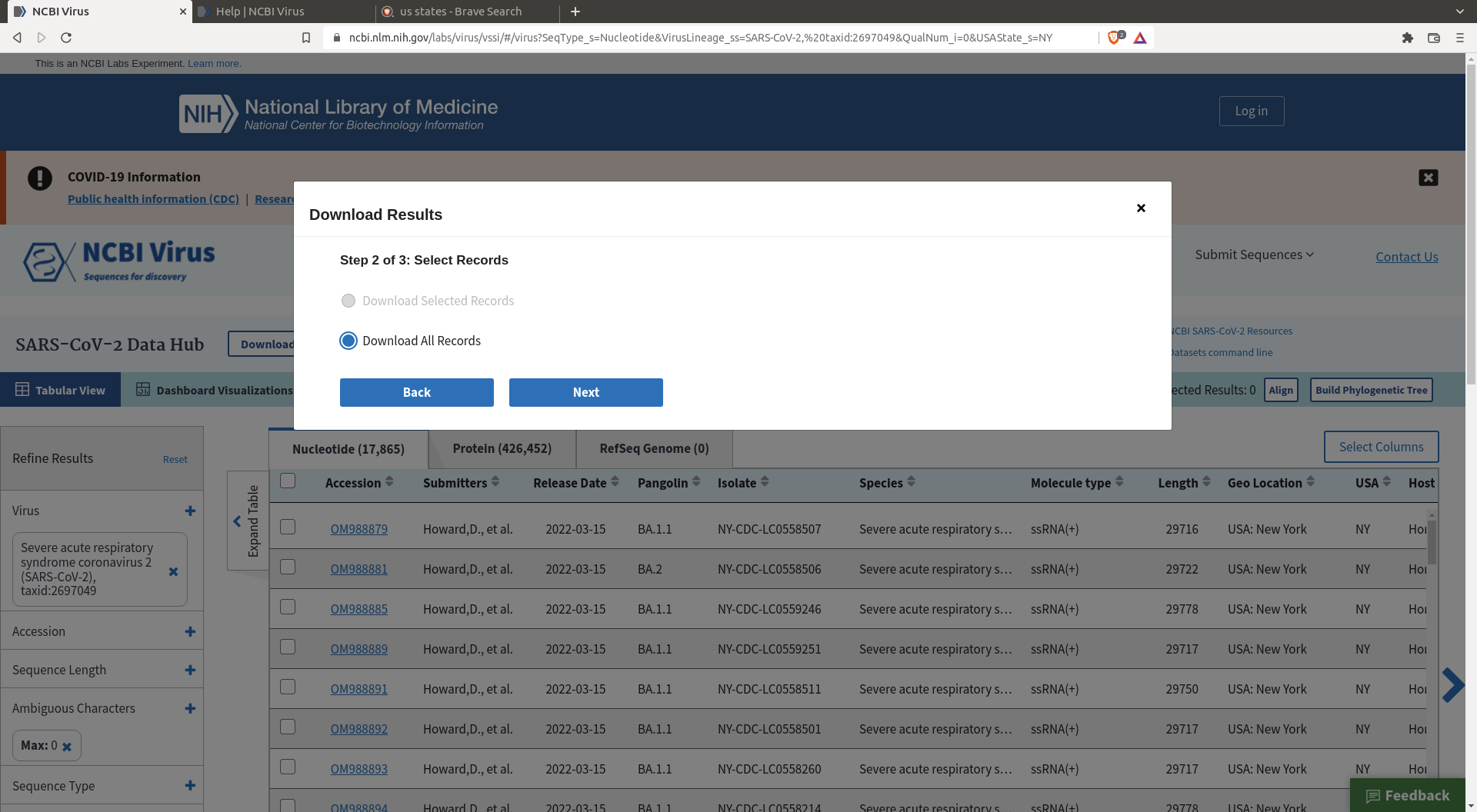
Then click download, specify the size of the random sample, and define the header you want for your sequences.
One also needs a reference sequence for the future alignment : https://www.ncbi.nlm.nih.gov/nuccore/MN908947.3/
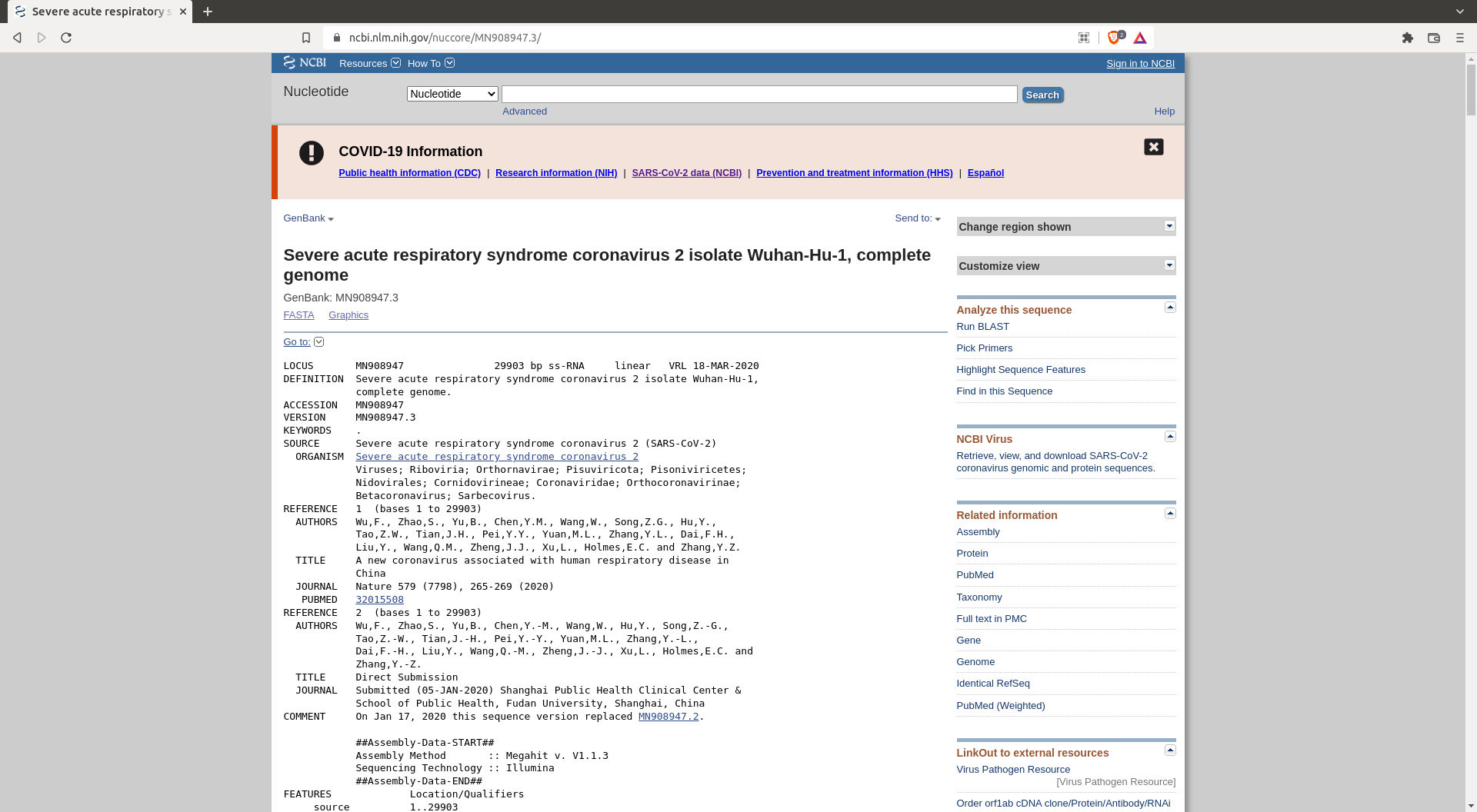 Take this sequence and save it as
Take this sequence and save it as reference.gb in your data folder.
Curating the dataset
Look at what you get on Aliview or similar. It's around 500MB. Make sure the sequence names contain the relevant information.
Building the pipeline
The steps are very similar to the Zika tutorial, except that we must create the metadata file from the sequence names and change the parameters in the filter part to correctly subsample the dataset. You need this subsampling step otherwise it the following steps will take a long time to run (too many sequences).
We suggest putting the raw data and everything you want to keep in a folder called data. The files that you want to clean from one analysis to the next will be in another folders that will be removed via a snakemake clean rule.
Now it is time to create the snakemake pipeline. You can inspire yourself from the zika tutorial as it will be very similar.
First define the input files you are going to use:
INPUT_FASTA = "data/SC2_sequences.fasta"
REFERENCE = "data/reference.gb"
DROPPED = "data/dropped.txt"
AUSPICE_CONFIG = "auspice_config.json"
You need to create the dropped.txt file even if it stays empty.
Also define the cleaning rule:
rule clean:
message: "Removing directories: {params}"
params:
"results",
"visualisation"
shell:
"rm -rfv {params}"
Metadata
The metadata concerning our sequence is contained in their headers. To create the metadata from these headers one can use the augur parse command.
A snakemake rule can be created for this, for example:
rule metadata:
message:
"""
Creating the metadata file from raw sequences {input.sequences}
"""
input:
sequences = INPUT_FASTA
output:
metadata = "data/metadata.tsv",
sequences = "data/sequences.fasta"
params:
fields= "accession country date"
shell:
"""
augur parse \
--sequences {input.sequences} \
--output-sequences {output.sequences} \
--output-metadata {output.metadata} \
--fields {params.fields}
"""