In a new preprint by Trevor Bedford, Rodney Daniels, Colin Russell, Boris Shraiman, and myself, we show how antigenic evolution can be integrated with the phylogenetic tree of HA sequences. Human immunity is the main driving force of the seasonal influenza viruses. Only when changing their antigenic properties, influenza viruses are able to re-infect previously infected humans. Mutation that prevent immune recognition often rapidly spread across the globe. Human antibodies typically recognize the tip of the HA trimer on the surface of the influenza virus and the amino acids at those positions change very often through time.
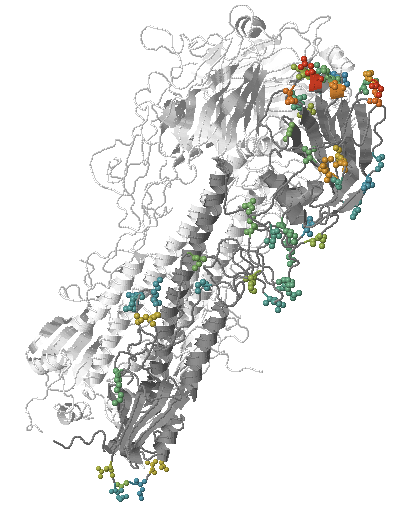
Mutations with antigenic effects highlighted on a HA structure
The antigenic properties of circulating influenza viruses are constantly monitored by the national influenza centres and the WHO collaborating centres for influenza using hemagglutination inhibition assays. These assays basically record how much an antiserum (obtained from a ferret) can be diluted and still recognise a virus -- if the virus has changed antigenically relative to the serum, even high serum concentrations don't inhibit the virus.
Results from such assays come in large tables of numbers quite removed from the molecular evolution of the HA protein. In the preprint, we show that antigenic distances behave similarly to sequence distances on the tree and can be explained as a sum of contributions associated with amino acid substitutions or contributions on branches of the tree.
To visualize antigenic evolution, we integrated HI titer data (mostly from the Worldwide Influenza Centre; in London and was generated by John McCauley, Rodney Daniels, and colleages) into nextflu. The site allows to selected a particular reference virus or vaccine strain and will color all viruses on the tree according to their measured or predicted antigenic similarity to the reference virus, see screen shot below. This integration makes it easy to associate antigenic changes with genotypic changes and the dynamics of the corresponding clades.
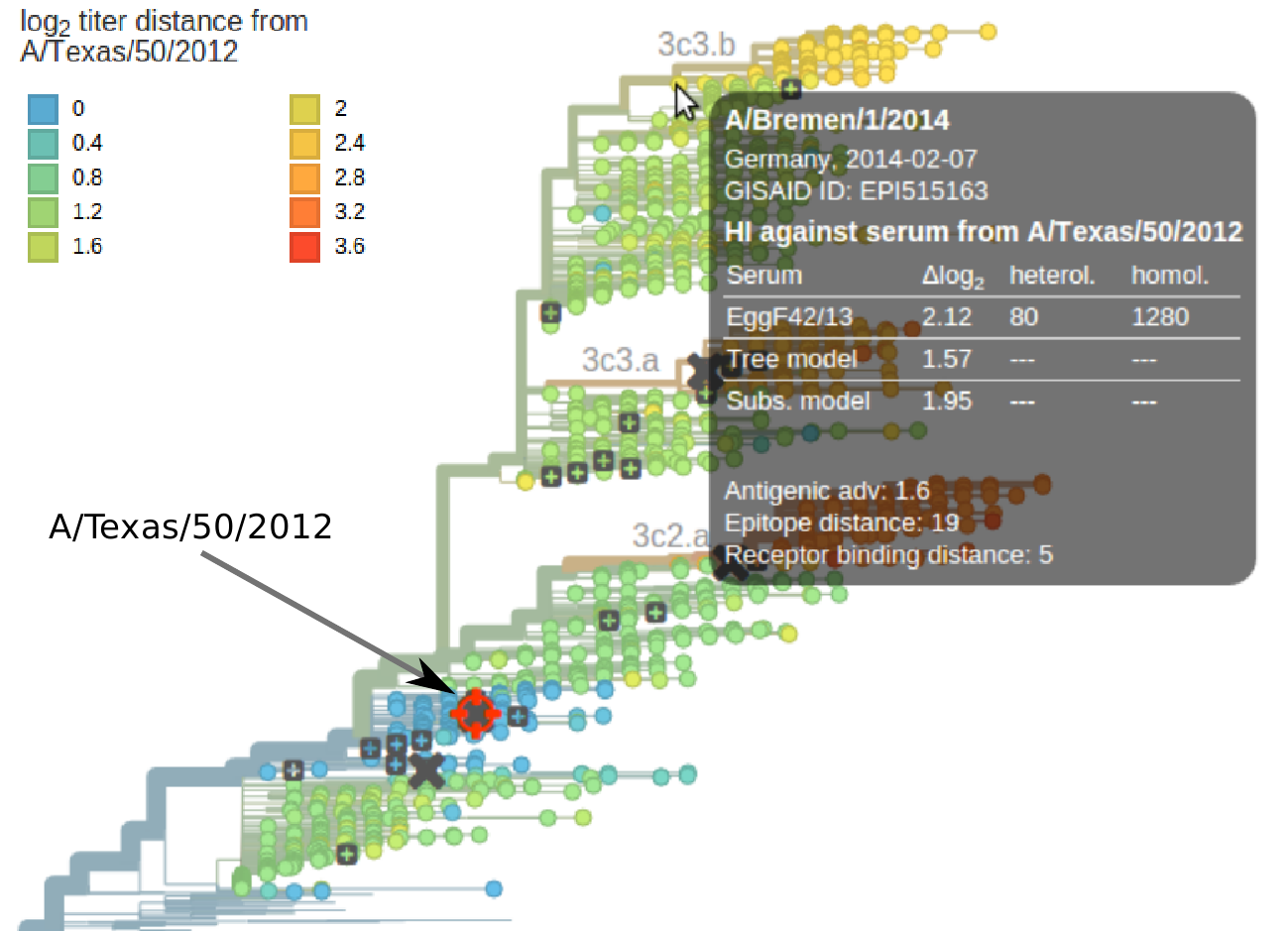
In the manuscript, we further investigate to what extent antigenic changes are predictive of the composition of future influenza populations. Successful clades tend to be antigenically advanced, but a substantial fraction of antigenic advances fail to spread. The appeal of HI data in the context of prediction is the early detection of a novel variant that is suboptimally recognized by the vaccine. However, many such "hopefuls" often go extinct and reliable prediction using HI data has find ways to differentiate antigenic changes that are likely to be successful from those that compromise virus functionality.